基于改进TF-IDF算法的基因通路富集方法
2022-10-09徐淑坦冷银辉陈明
徐淑坦,冷银辉,陈明
1.上海海洋大学信息学院,上海 201306;2.农业农村部渔业信息重点实验室,上海 201306
前言
在生物医学相关研究领域,随着高通量测序技术的发展,组学数据的规模也呈指数级增长。从庞大的组学数据中,可以利用生物信息学技术挖掘与疾病发生机制相关的通路,对疾病的诊断和治疗具有重要意义。
在过去10 多年,已经开发了很多基因功能富集分析方法来识别各种疾病相关的通路[1-3]。基于数据来源和算法大致可以将基因功能富集分析方法分为4 大类:过代表分析(Over-Representation Analysis,ORA)、功能集打分(Functional Class Scoring, FCS)、基于通路拓扑结构(Pathway Topology,PT)和基于网络拓扑结构(Network Topology,NT)的方法[3]。ORA方法首先选定一组感兴趣的基因作为基因列表,然后对该基因列表与通路中的基因集做交集,找出它们共同的基因并进行计数,最后利用统计检验的方式来评估计数值是否显著高于随机,即待测通路在基因列表中是否显著富集。基因集富集分析(Gene Set Enrichment Analysis, GSEA)属于FCS 类方法,是最具代表性的基因功能富集分析方法,该方法利用基因表达和表型数据对所有基因进行排序,然后计算基因集Kolmogorov-Smirnov(KS)统计量,即在基因排序列表中靠近两端程度的得分,最后通过置换方法评估基因集的显著性[4]。FCS 类方法将通路中的基因视作独立个体,实际上通路中的基因通过复杂的相互作用来影响细胞的发育、分化或疾病等生物学过程[5]。之后一些基于PT 的方法开始考虑通路的信息,Liu 等[6]提出一种基于定向随机游走的方法来推断通路活性,即利用基因在定向通路中的结构信息来评估每个基因在通路中的重要性,然后使用重要性对基因加权进行分析。Deng 等[7]利用蛋白质互作数据和通路的基因集构建了蛋白质-通路交互网络,从全局层面优化富集分析。Yang等[8]提出一种基于PT 的通路富集方法,该方法根据基因节点的全局上游或下游位置和通路中的连接度数来评估节点的重要性,富集出那些拥有更多在上游或中枢节点中的基因的通路。后来的基于NT 的方法中,很多方法也借鉴了GSEA 的思想。Winterhalter 等[9]提出基于蛋白质互作网络的GSEA 方法JEPETTO,利用网络的拓扑结构信息分析基因和通路之间的功能关联。Rahmati等[10]整合来自20个通路数据库的核心通路,构建了庞大的蛋白质互作网络,以求富集出与生物学功能相关的通路,提出pathDIP 方法。Han 等[11]提出基于人类基因功能网络的GSEA 方法NGSEA,该方法衡量基因集的富集分数,不仅考虑单个基因的表达差异,还考虑功能网络中它们的相邻基因的表达差异。Yoon 等[12]提出一种新的网络加权的GSEA,对基因集进行富集分析时,将重叠基因和蛋白质互作网络结合起来,有效地识别出与基因功能相关的基因集。Zito 等[13]将图论中衡量结点的中介中心性权重引入通路富集分析,利用蛋白质互作网络,提出网络节点中心性加权的GSEA。从更高层面看,通路是生物互作网络的一部分,因此通路富集分析有必要综合考虑基因在通路的局部信息和在通路数据库的全局信息,这一点和数据挖掘领域的常用加权算法TF-IDF(Term Frequency-Inverse Document Frequency)的思想类似,该算法综合考虑字词在文件的局部重要性和文件库的全局特异性两方面来评估字词的权重。
针对以上问题,本研究融合基因在通路的局部重要性和在通路数据库的全局特异性定义基因影响力,提出一种基于改进TF-IDF 算法的基因通路富集方法(GIGSEA 方法)。首先利用基因相互作用数据来计算通路基因的影响力,然后通过基因表达数据和表型数据来计算基因与表型的相关性值,并利用基因影响力和相关性值计算富集分数,最后通过统计学方法计算通路的显著性P值。在肝细胞癌(Hepatocellular Carcinoma, HCC)和结肠直肠癌(Colorectal Cancer, CRC)数据集的实验结果表明该方法能有效识别出与疾病相关的通路,对于今后研究疾病的发生发展机制有重要的指导意义。
1 材料与方法
1.1 数据集及预处理
本研究的通路数据来自KEGG 数据库[14],KEGG数据库是代谢组学和蛋白质组学研究中常用的生化通路数据库,从GSEA 网站下载通路数据集,共包括186个通路,共有5 245个基因。
本研究的基因相互作用数据从STRING 数据库[15]中获得,下载地址为https://www.string-db.org/cgi/download,下载的数据是最新的11 版本的人类蛋白互作数据,共11 938 498 对,从中提取包含KEGG通路基因的基因相互作用,去除重复的相互作用对,共计977 800对。
本研究的基因表达数据包括HCC 和CRC 数据集。HCC 数据集来自TCGA 数据库,包括来自肝癌患者的肿瘤邻近组织的50 对配对样本和324 个肿瘤样本,本研究只使用50对配对样本进行测试,即包括50 个肿瘤样本和50 个正常样本,该数据集下载自UCSC Xena 网站(https://xena.ucsc.edu/)[16]。CRC 数据集来自GEO(Gene Expression Omnibus)数据库的GSE8671 表达谱数据,包括32 个结肠癌腺瘤组织和32个配对的癌旁组织。
1.2 方法
1.2.1 基于改进TF-IDF 算法计算基因影响力在TFIDF 算法中,TF 是词频(Term Frequency),即词条t 在文件中出现的频率,TF 越大,意味着t 可能是文件的关键词,说明t 越重要;IDF 是逆文件频率指数(Inverse Document Frequency),如果包含词条t 的文件越少,IDF 越大,说明t 具有很好的类别区分能力,t的权重相应地越大。TF是对词条t在本文件的评估,IDF是对t在文件库的评估。
类似地,本研究的基因影响力由基因在本通路的局部重要性和在通路数据库的全局特异性来衡量。通路基因的相互作用的关系可以抽象为一个图,本研究定义基因的重要性为在通路中与其他基因产生相互作用的数量,即在通路图中的连接度。采用基因频率GF(Gene Frequency)来表示基因重要性,GF表示为:

其中,ni,j是基因j在通路pi中的度数,分母则是通路pi中所有基因的度总和。
一个基因的特异性表现为基因在所有通路中出现的频度,频繁出现在很多通路中的基因,它们对通路的影响相对较小;仅在少数通路中出现的基因其特异性高,它们的差异表达对通路的影响就大。本研究定义逆通路频率(Inverse Pathway Frequency,IPF)来表示基因特异性,IPF表示为:
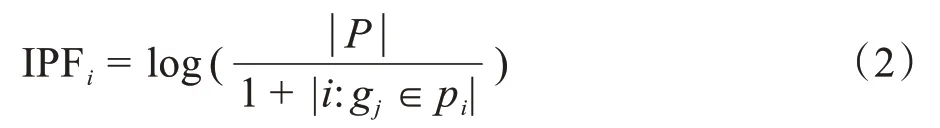
其中,|P|是数据库中的基因通路的总数;|i:gj∈pi|表示包含基因gj的通路数目,即ni,j≠0的通路数目。
某基因在特定通路内的重要性越高,并且在通路数据库中的特异性越高,那么该基因影响力越大。合并GF和IPF计算基因影响力GI(gi,j),表示为:

其中,GI(gi,j)表示基因j对通路i的影响力大小。
1.2.2 GIGSEA 方法假定有N个基因、T个样本的基因表达数据,T个样本包括两种表型pos 和neg,样本数量分别为t1 和t2;给定一个通路pi,包括Q个基因。GIGSEA方法主要过程如下:
(1)利用基因相互作用数据统计每个基因在通路中的连接度数量,计算出GF(图1a)。然后统计每个基因在全部通路中出现的频数,计算出基因IPF。最后合并GF 和IPF计算GI(gi,j)。
(2)考虑到通路中有一些基因不在基因表达数据的基因集中,对两个集合取交集,计算交集内的基因与表型的相关性值,计算公式为:

其中,函数mean(x)、std(x)分别指表型x的基因表达值的平均值和标准差。最后按相关性值从大到小排序得到列表L=[[g1,r1],…,[gj,rj],…,[gm,rm]],包括M个基因(图1b)。
(3)计算通路的富集分数。从列表L 的第一个基因开始,当遇到一个在通路pi里面的基因(hits),则增加分数;遇到一个不在pi里面的基因(misses),则减少分数,具体公式为:
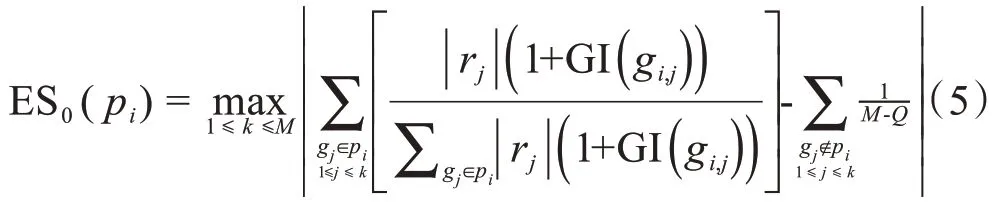
最终得到一个分数曲线(图1c),曲线上的点到横坐标距离的最大值即为ES0(pi)。
(4)随机置换基因Nperm次(实验中,Nperm取基因富集分析常用的值,1 000次)。随机置换基因指随机在列表L 挑选Q个基因作为通路基因。重复步骤(3),计算置换后的通路富集分数ESperm(pi)。如图1d所示,最后统计|ESperm(pi)|>|ES0(pi)|的数量Nsign,P值等于Nsign与Nperm的比值。

图1 GIGSEA方法流程图Figure 1 GIGSEA flowchart
为了方便比较,本研究按GSEA 方法[4]计算校正后的富集分数(Normalized Enrichment Score, NES)和错误发现率(False Discovery Rate,FDR)。
1.3 结果评价
本研究将显著性P值、FDR 和|NES|的阈值分别设为0.05、0.25 和1.00,筛选出具有显著性意义的通路,为了检验方法的有效性,与GSEA方法进行比较。两种方法的显著通路大部分是重叠的,重叠通路只是排名的略微差异,因此关注显著通路的差集更有意义,将显著通路差集内的通路定义为差异通路。本研究主要从差异通路的3 个方面来说明方法的有效性,包括(1)生物学解释验证。通过查阅关于HCC或CRC 相关的生物学研究文献来验证通路与HCC或CRC 存在的某种联系。大部分富集方法都通过这种方式来解释,如果通路中的某些基因或者产物对疾病产生影响,那么该通路与疾病是相关的。(2)相关文献数量。通过PubMed 生物医学论文数据库检索通路和HCC 或CRC 存在联系的文献,利用文献数量的多少来表示差异通路与疾病相关性的强弱。如果检索词全部出现在文献中,那么该条文献会出现在PubMed 检索结果中,因此文献数量从一定程度上可以反映通路与疾病的相关性。比如对于Jak Stat Signaling Pathway 与HCC 的相关性,通过关键词Jak Stat、Hepatocellular Carcinoma 来进行搜索,排除掉Signaling Pathway 这些冗余的词;而对于Asthma 和Peroxisome 通路,则采取补全关键词Pathway 来检索。(3)通路基因集对应的表达数据对表型的分类性能。每一个通路基因集对应的基因表达数据和样本表型数据输入到构建的支持向量机(Support Vector Machine,SVM)分类模型中,求出AUC,结合计算的P值、FDR 和||NES ,利用分类性能对两种方法进行有效性比较[17]。AUC 是分类模型中常用的评价指标,其取值范围为0.5~1.0,AUC 越大,分类效果越好,意味着通路基因的表达值对疾病分类越准确。比如HCC 数据集包括100 个样本,有Normal 和Cancer 两种表型,各50个;而通路Jak Stat Signaling Pathway中有155 个基因,则输入SVM 模型的数据为:100 个样本,每个样本有155 个特征,对应155 个基因,每一个样本的标签对应于表型,比如Normal 为0、Cancer 为1。因此这是一个针对样本表型的二分类问题,如果两种表型的基因表达值存在的差异更高,则更能够把表型区别开来,产生更高的AUC,意味着通路内的基因和HCC疾病的相关性更高。
2 结果与讨论
2.1 HCC数据集的结果与讨论
HCC 数据集的富集结果如表1 所示,表中共有33个通路,为两种方法显著通路的并集。GSEA 富集出30 条通路,GIGSEA 富集出29 条通路,且富集出3个新的通路:Jak Stat 信号通路(Jak Stat Signaling Pathway)、过氧化物酶体通路(Peroxisome)、半胱氨酸和蛋氨酸代谢通路(Cysteine and Methionine Metabolism),分别排在第5、8、11 位,与GSEA 相比,它们的排名也都提高了。

表1 HCC数据集中两种方法的富集结果Table 1 Enrichment results of two methods in HCC dataset
经过查阅文献发现,GIGSEA 的差异通路都与HCC疾病的发生机制有一定的联系。Jak Stat信号通路的失调可能导致包括HCC 在内的各种癌症[18]。Tang 等[19]发现Jak Stat 信号通路在HCC 中维持具有肿瘤增殖能力的癌症干细胞以及创建免疫抑制微环境,该通路中的STAT3 基因对靶基因和蛋白质的调节促成肿瘤发生的概率。目前针对HCC,已经开发出多种JAK 或STAT 小分子抑制剂和RNA 疗法。过氧化物酶体通路中,过氧化物酶体包含至少50 种不同的酶[20]。Xu等[21]发现过氧化物酶体增殖物激活受体δ(PPARδ)是一种核转录因子,与肿瘤发生有关,通过PPARδ 和前列腺素信号通路之间的串扰,两个通路共同调节人体HCC 细胞的生长。Wirtz 等[22]及Zhuang 等[23]发现HCC 细胞转移与半胱氨酸和甲硫氨酸代谢通路中代谢物水平和代谢酶表达的改变有关,该通路的ENOPH1 的过表达促进细胞迁移和侵袭,而ENOPH1 下调抑制细胞迁移和侵袭,研究表明ENOPH1 可以促进HCC 进展,可以作为HCC 的生物标志物和治疗靶点。
相关文献可以反映已有研究对于通路和疾病具有相关性的支持,因此本研究对两种方法的差异通路进行相关文献统计,验证其与HCC 的相关性。统计结果如表2 中文献数量一列所示,GIGSEA 和GSEA 的差异通路的文献数量均值分别为129 和6,GIGSEA 明显高于GSEA,表明GIGSEA 的差异通路与HCC的相关性是更高的。
接着利用SVM 模型来测试差异通路对HCC 数据集的两种表型的分类性能。GIGSEA 的平均P值和平均FDR 远小于GSEA,且||NES 大于GSEA,从统计学上表明GIGSEA 差异通路与HCC 相关性更强。两种方法的差异通路在SVM模型都表现出良好的分类效果,GIGSEA 和GSEA 的AUC 分别达到99.22%和96.99%,表明GIGSEA 差异通路的分类性能同样优于GSEA差异通路(表2)。

表2 HCC数据集差异通路的对比Table 2 Comparison of the differential pathways in HCC dataset
2.2 CRC数据集的结果与讨论
CRC 数据集的富集结果如表3 所示,表中共有26 个通路,包括GIGSEA 和GSEA 的显著通路。GSEA 富集出20 条通路,GIGSEA 富集出23 条通路,且富集出6个新的通路:肌动蛋白细胞骨架的调节通路(Regulation of Actin Cytoskeleton)、白细胞跨内皮迁移通路(Leukocyte Transendothelial Migration)、补体和凝血级联通路(Complement and Coagulation Cascades)、朊病毒病通路(Prion Diseases)、哮喘通路(Asthma)、肾素血管紧张素系统通路(Renin Angiotensin System),它们的排名在GIGSEA 中都有一定的提高。

表3 CRC数据集中两种方法的富集结果Table 3 Enrichment results of two methods in CRC dataset
文献检索显示除了哮喘通路,GIGSEA 的差异通路都与CRC 有一定的联系。Kanaan 等[24]发现肌动蛋白细胞骨架调节通路通过细胞骨架蛋白,如Fascin-1,参与转移性散发性CRC 的发展;与散发性CRC 相比,该通路的调控基因之间的相似遗传多态性和突变也可能与CRC 的发育不良、癌变以及侵袭和转移的易感性增加有关。Tremblay 等[25]发现E-选
择蛋白被结肠癌细胞激活会触发p38和ERK MAP激酶的激活,从而诱导细胞骨架重塑,导致内皮层破裂,促进粘附的结肠癌细胞的外渗,该研究表明E-选择蛋白介导的p38和ERK MAP激酶激活对结肠癌细胞跨内皮迁移的调节。Matilda等[26]发现补体和凝血级联通路、参与脂质代谢的通路、急性期反应信号通路是高C 反应蛋白(C-reactive Protein, CRP)CRC 患者的主要干扰通路。细胞朊病毒蛋白(Cellular Prion Protein,PrPc)是一种细胞表面蛋白,由朊病毒通路中的PRNP 基因编码[27]。Ong 等[28]发现PrPc 的过表达可能通过诱导内皮增殖-分化开关的方式参与CRC诱导的血管生成。Chen等[29]发现肾素血管紧张素系统抑制剂的使用与CRC 风险和死亡率降低有关,该研究通过实验证明肾素血管紧张素系统抑制剂使用持续时间每增加一年,CRC风险降低6%。
同样地,通过Pubmed检索和SVM模型分类来验证差异通路与CRC 之间的相关性,结果如表4 所示。GIGSEA 差异通路的相关文献数量的均值为55,而GSEA 为34;GSEA 的P值要小于GIGSEA,两种方法的差异通路平均FDR 和||NES 都接近;GIGSEA 的平均AUC 明显高于GSEA,分别为91.32%和85.67%,表明GIGSEA 的差异通路的分类性能优于GSEA。综合考虑,GIGSEA 的差异通路与CRC 的相关性比GSEA强。
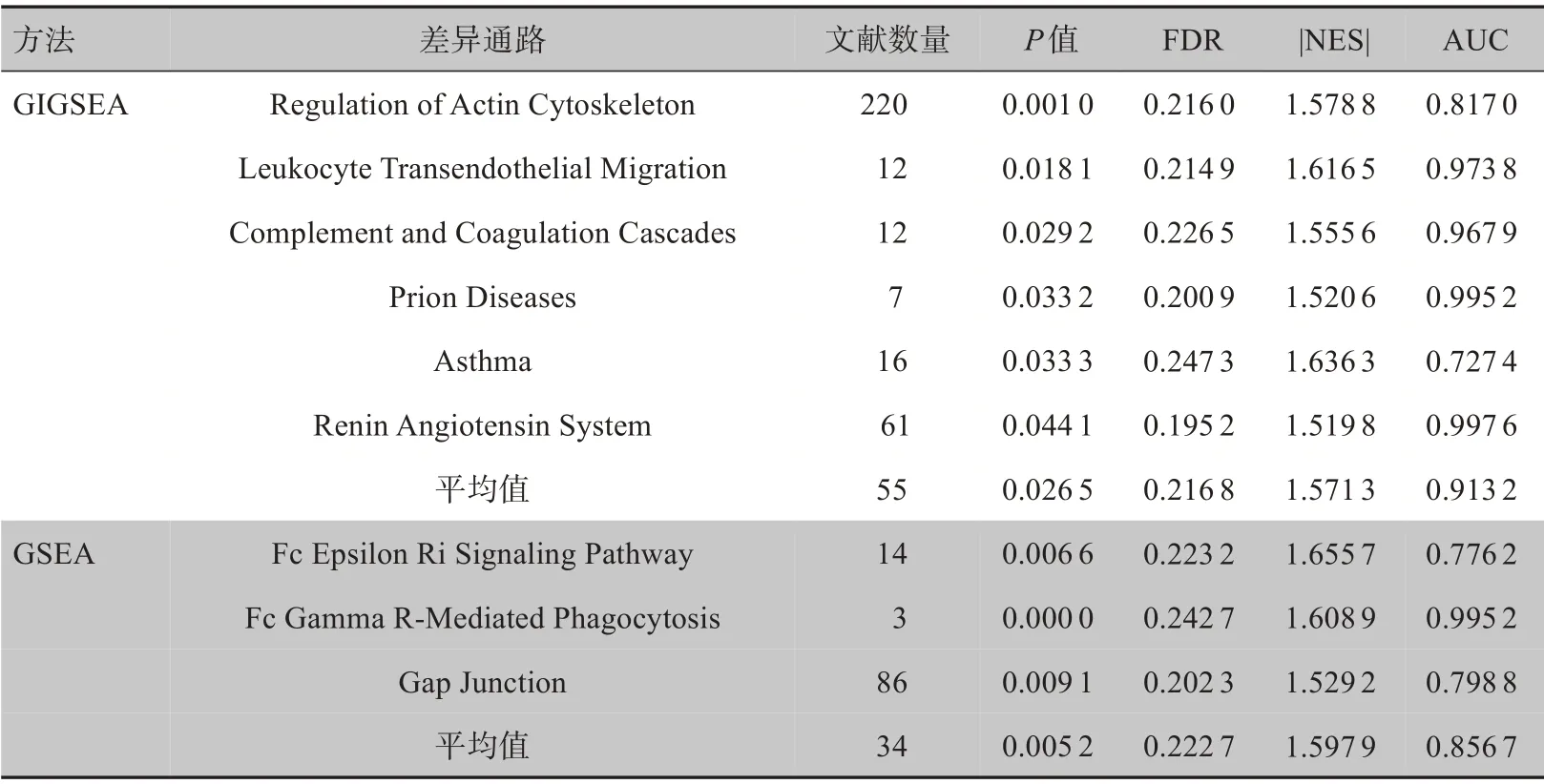
表4 CRC数据集差异通路的结果对比Table 4 Comparison of the differential pathwaysin CRC dataset
2.3 基因通路富集分析网站
为了方便用户使用GIGSEA进行通路富集分析,本研究基于SSM框架开发了一个在线的基因通路富集分析的网站,SSM 框架是Java 企业级开发领域Spring、Spring MVC 和MyBatis 框架的缩写。本研究利用bootstrap-tablejs组件来绘制富集分析的结果展示页面,可以对富集结果进行排序、搜索等功能(图2)。
图2 富集结果展示页面Figure 2 Enrichment result display interface
本研究利用EChartsjs 组件的关系图来进行基因通路的可视化,如图3 所示,通路图中的节点表示基因,节点之间的连线表示基因之间的相互作用关系,基因节点的大小与基因相关性值的绝对值成比例,基因节点的颜色与基因列表相关性值正负相关,红色代表相关性值为正,蓝色代表相关性值为负,灰色代表该基因不在基因列表中。

图3 Echart可视化的通路局部示意图Figure 3 Partial schematic diagram of Echart visualized pathways
3 结论
本研究提出一种基于改进TF-IDF 算法的GIGSEA 方法。首先利用通路基因相互作用数据,考虑基因在通路的局部重要性和在通路数据库的全局特异性,计算基因的影响力;然后利用基因表达数据和表型数据计算基因与表型的相关性值;接着融合基因影响力和表型相关性值,计算通路的富集分数;最后通过置换基因的方式,考察通路是否和疾病相关。本研究利用HCC和CRC数据集来测试GIGSEA的效果。与GSEA 比较,本研究发现了与HCC 相关的3个新通路,以及与CRC 相关的6个新通路。除了哮喘通路,本研究都找到研究文献来证实通路与疾病之间的相关性。利用PubMed 检索相关文献的结果显示在两个数据集中,GIGSEA 的文献数量都远远多于GSEA。利用SVM 模型分类的结果显示在两个数据集中,GIGSEA 通路对应的表达数据的分类效果都优于GSEA。GIGSEA 方法不仅丰富了富集分析方法,更重要的是为发现与疾病相关的通路提供了一种新思路。
