基于单畴表征的高/低黏滞磷脂膜中的相分离*
2022-09-30朱玉洁朱涛盛洁周琪蒋中英
朱玉洁 朱涛 盛洁 周琪 蒋中英‡
1) (伊犁师范大学电子与工程学院,微纳电传感技术与仿生器械重点实验室,伊宁 835000)
2) (南京大学物理学院,固体微结构物理国家重点实验室,南京 210093)
磷脂相分离是细胞脂质筏形成的物理驱动力,在生命物质的空间组装中发挥着重要的作用.本研究通过单微畴跟踪、径向波动性分析等手段定量地研究了多组分磷脂相分离动力学.发现在低线张力差异下,大相的黏滞性是产生微畴粗化差异的主要原因.融合产生的流场促进微畴扩散,加速了低黏滞大相中微畴的融合粗化;而高黏滞大相中微畴主要依赖布朗运动扩散,融合粗化较慢.进一步建立微畴的扩散与融合粗化理论模型,理解了大相黏滞性较高与较低时,微畴尺寸与粗化时间分别满足的0.5 与1 幂指数关系.此外还发现,可以通过胆固醇相对含量调节大相黏滞性,提高了微畴粗化的可控性.研究深化了多组分磷脂相分离机制的理解,为调控细胞膜表面的生物分子再分布提供了有价值的参考.
1 引言
细胞膜表面的生物分子空间组装对生命功能至关重要.在二维的细胞膜中,脂质筏将不同性质的磷脂和蛋白分子在侧向分拣成高有序并相对稳定的结构(富饱和磷脂与胆固醇)[1,2].在信号传导、蛋白输运等诸多涉及膜内分子组装的过程中,脂质筏发挥了高效浓缩特异性分子的作用[3-5].
研究发现,可以通过磷脂间相互作用引起侧向组分相分离,形成与脂质筏组分相近的结构[6].因此,多组分巨单层囊泡(GUVs)被用于研究脂质筏的物理化学性质.研究发现,相分离的热力学性质(如相分离的形成条件及稳定性)由一系列实验参量(如温度[7,8]、混合磷脂比例[9]等)决定.在高温下,磷脂的混合熵较高,GUVs 形成单一的液相;降温导致熵降低,不同磷脂间的焓作用差异导致GUVs 形成排布有序和无序的相分离结构[7].
由于细胞不是一个封闭的热力学平衡体系[10],研究多组分GUVs 相分离的形成动力学对理解脂质筏的动态组装与调控是至关重要的[11].国内外众多课题组立足生物分子组装开展了系统的实验与理论研究[12,13].Stanich 等[14]报道,降温导致GUVs表面形成了许多纳米尺度的微小圆形微畴.微畴可通过两种机制粗化: 一是融合粗化,即微畴通过扩散与周边的微畴碰撞、融合形成更大的微畴;二是蒸发-凝聚粗化,磷脂从较小的微畴边界蒸发并结合到更大的微畴边界.并且,微畴的粗化动力学受到多种因素的影响.Garcia-Saez 等[15]发现,不同组分间的线张力驱动着微畴的粗化,以降低微畴的总边长.提高线张力可显著加快微畴的粗化.Rozovsky 等[16]发现,在线张力的驱动下,过量膜面积可诱导产生微畴出芽.芽颈的膜曲率产生微畴间的近程排斥,使得融合粗化动力学受阻.Li 等[17]进一步给出,当线张力超过临界值,芽结构可与母体完全分裂.增大自发曲率可降低该临界线张力值,并提高微畴分裂的频率.此外,降温速率控制的成核点数量等也会影响微畴的粗化动力学[14].但以上研究往往聚焦较高线张力下的微畴粗化动力学.在细胞脂质筏涉及的低线张力条件下,相分离的产生及其影响机制尚待深入研究.
最近一些课题组将单微畴表征策略应用于微畴粗化与组装研究.Talbot 等[18]采用激光共聚焦显微三维(3D)重构了单个微畴在温度梯度场中的运动,发现了线张力梯度引起的异常微畴超扩散,给出了微畴种类无关的、在高温侧融合粗化的内在机制.Wongsirojkul 等[19]采用宽场荧光显微追踪了单个微畴的径向波动,发现了膜张力产生的高线张力,给出了组分依赖的、低渗条件下磷脂膜相分离机制.通过单微畴的轨迹跟踪、径向波动分析等,可以定量地给出线张力、侧向扩散等膜性质,从而为磷脂相分离研究提供重要的分析数据.
本文采用单微畴表征策略研究了类细胞脂筏的产生与调控.研究采用了较高的实验温度以产生较低的线张力;选择了4 个线张力差异较低的组分,考察了它们的微畴粗化动力学.发现可以通过大相黏滞性调控微畴的尺寸.给出了不同黏滞性下的微畴扩散与粗化机制差异,获得了大相黏滞性决定的微畴尺寸与时间标度关系.并发现可以通过胆固醇调控大相黏滞性,阐明了胆固醇相对含量反向影响液态有序、无序相黏滞性的机制.
2 实验
2.1 电方法制备巨型单层囊泡(GUVs)
采用电形成方法制备GUVs[20].二油酰磷脂酰胆碱(DOPC)、二棕榈酰磷脂酰胆碱(DPPC)、胆固醇(Chol)和1,2-二油酰-SN-甘油—3-磷酰乙醇胺(Rhod-PE)购于Avanti Polar Lipids 公司.一般地,将给定组分的磷脂溶于氯仿中,掺杂摩尔分数为0.5% 的Rhod-PE.取6 µL 磷脂溶液涂抹至ITO 玻璃导电面(电阻率为 5×10-4Ω·cm),真空干燥2 h.在两块ITO 玻璃中夹放0.3 mm 硅胶垫片形成GUVs 制备舱室.将300 mmol/L 蔗糖溶液缓慢注入舱内.施加峰峰值5 V、频率10 Hz 的正弦交流电,温度设置为60 ℃培养3 h 获得GUVs.
2.2 荧光显微镜表征三组分磷脂相分离及相图
采用Olympus IX7 倒置荧光显微镜(配置Andor 897 EMCCD、532 nm 激光光源、100×油镜)观察GUVs 表面的磷脂相分离.选用的GUVs直径在20—50 µm,将GUVs 静置于两片玻片之间硅胶涂制的舱室里.在观测前,电形成的GUVs被稀释于295 mmol/L 葡萄糖溶液中.GUVs 内部的渗透压略高于外部,低正张力膜可消除过量的膜面积,并且内外溶液密度差使GUVs 沉淀至物镜侧玻片表面以便于观测[21].GUVs 温度通过Warner显微镜热台进行控制(控温精度0.1 ℃).一般地,从42 ℃ (混溶性转变温度以上)快速降温至32 ℃(混溶性转变温度以下)以形成磷脂相分离结构(—10 °C/min),并在温度稳定在32 °C 后再进行显微镜观测.微畴的平均半径(r)通过灰度阈值确定的面积计算获得.统计使用的微畴中心处于GUVs中3/10 囊泡直径划定的圆形范围内,以降低二维投影分析产生的误差.微畴的面密度通过观测半球的微畴数量除以半球表面积获得.实验获得的DOPC/DPPC/Chol 三组分相图与之前报道的结果一致[22].基于三组分相图的等温连结线,获取了液态有序(Lo)与液态无序(Ld)畴中的磷脂组分.在相图中绘制Lo与Ld面积相同的组分连线,等温连结线近似于该线的垂线.等温连结线与Lo-Ld相分离相区边界的交点即为大相与微畴的组分.
2.3 线张力分析
线张力通过分析微畴的径向波动性获得[19].具体地,在温度降至32 ℃约5 min 后,采集数次约5 s 时长的微畴边界波动(6 帧/s).边界偏离半径的谱线以傅里叶级数展开的形式给出:

其中,ψ为相位角,k为波数,ak与bk为幅值.过剩自由能满足:
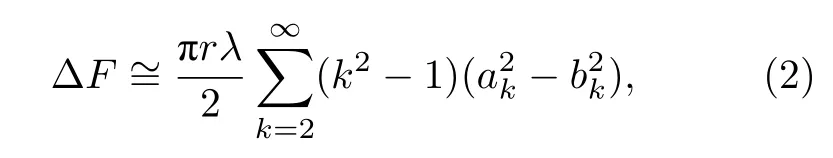
其中λ为线张力.根据能量均分定律,每个独立波数的自由能均为kBT[19].其中,kB为玻尔兹曼常数,T为绝对温度.因此,

线张力通过(3)式拟合微畴边界波动数据获得.选择较大GUVs (直径≥40 µm)中心的微畴用于线张力的计算.用于线张力分析的微畴仅受到微量的低强度激光辐照(总辐照时间短于10 s),以减小光氧化造成的线张力偏移.
2.4 微畴均方位移及侧向扩散速率分析
采集微畴的运动轨迹以分析其均方位移及侧向扩散速率[23].具体地,在温度降至32 ℃后约2—10 min 内,荧光显微采集约1 min 时长的微畴运动(6 帧/s).通过ImageJ 的TrackMate 模块重构微畴的扩散轨迹,以微畴中心在t时刻各方向的位移(x(t),y(t))计算均方位移(MSD(t)):

符合布朗扩散的微畴MSD(t)与t满足线性关系[24]:

其中D为侧向扩散速率.用于均方位移及侧向扩散速率分析的微畴满足以下要求: 1) 微畴的中心处于GUVs 中0.7 倍囊泡直径划定的圆形范围内,用以降低二维模型分析产生的误差[25];2) 微畴近圆形,其边缘到中心的距离处于0.85r—1.15r范围内.
2.5 膜黏滞性的荧光光谱分析
通过二苯基己三烯(DPH)荧光的各向异性表征磷脂膜的黏滞性[26].制备掺杂摩尔分数为0.5%DPH 给定组分的GUVs,使用Perkin Elmer LS55 荧光分光光度计获取发射光强.激发光波长设为360 nm,发射光波长设为430 nm.样品温度通过PTP1 温控组件(Perkin Elmer)恒定在32 ℃.DPH 的各向异性(a)使用下式计算:
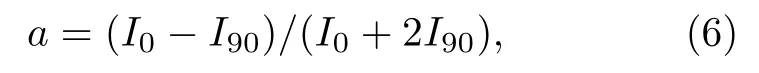
其中,I0和I90分别为平行、垂直于平面偏振激发光的发射光光强分量.a反映了磷脂膜的黏滞性.a越低则磷脂膜的黏滞性越低.
3 结果与讨论
首先,获取了DOPC/DPPC/Chol 三组分在32 ℃的相图(见图1(a)).在相图中的灰色区域,GUVs 形成液态有序(Lo)-液态无序(Ld)相分离.Lo相主要由分子排布较为紧密的DPPC 与Chol构成,而Ld相主要由分子排布较为疏松的DOPC构成[7].荧光染料Rhod-PE 选择性地混溶入Ld相[27],在532 nm 的激发下发出明亮的荧光.相图中的红色点线表示Lo与Ld面积相同的组分.在该点线的左侧,Lo微畴分散在Ld大相之中(如DOPC/DPPC/Chol 的摩尔百分比为27%/53%/20%,40%/40%/20%,分别简称为M1,M2).而在该线的右侧,Ld微畴分散在Lo大相中(如DOPC/DPPC/Chol 的摩尔百分比为11%/44%/45%,16%/64%/20%,分别简称为M3,M4).点线的垂直线近似为等温连结线[7].基于等温连结线与相区边界的交点,可获取大相与微畴的磷脂含量比例(表1).

表1 微畴与大相的混合磷脂含量Table 1.Lipid composition in domains and bulks.
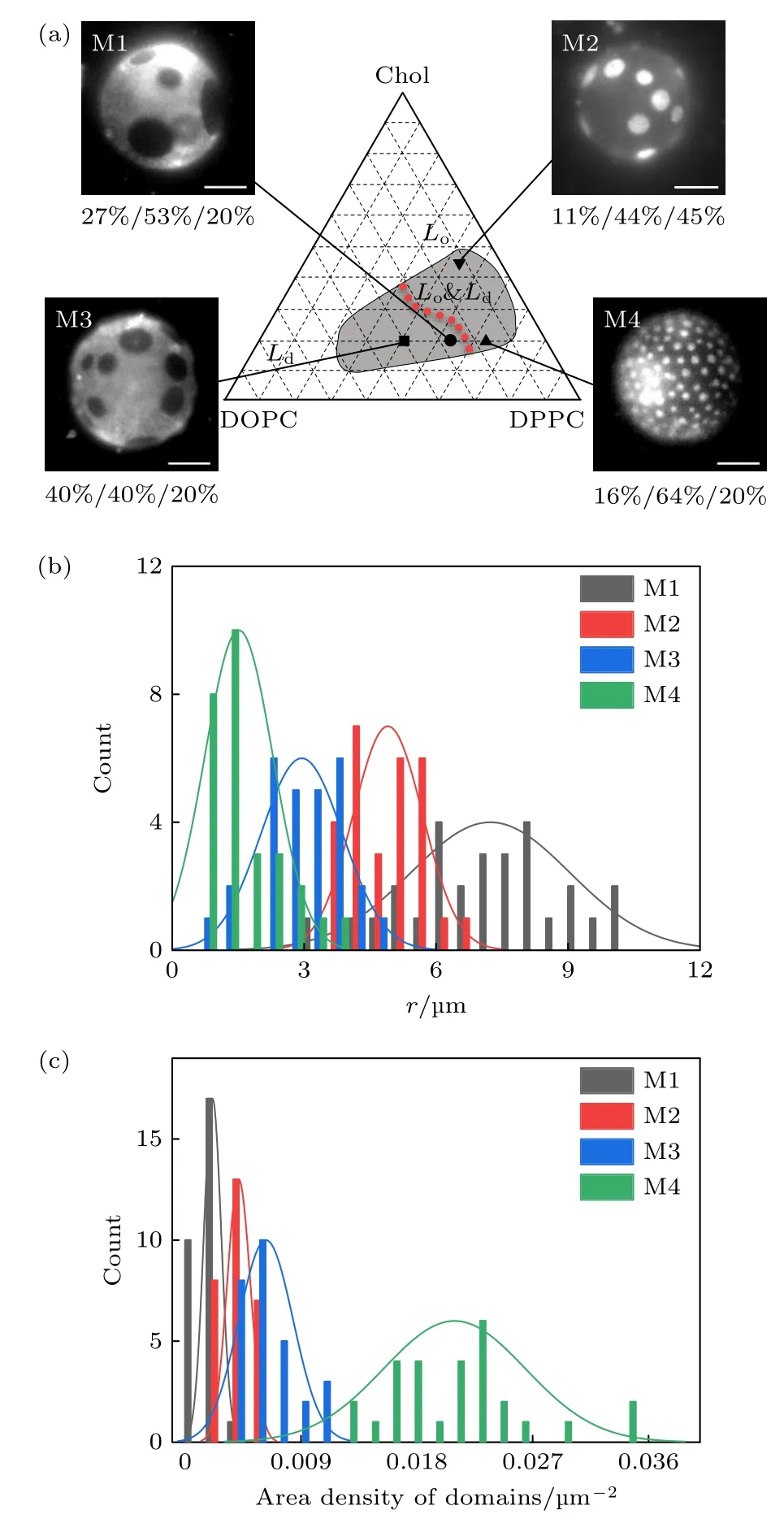
图1 DOPC/DPPC/Chol 三组分相分离 (a) 32 °C 的相图,红色点线表示Lo 与Ld 面积相同的组分,标尺为 10 µm;(b) 微畴尺寸分布图;(c) 微畴面密度分布图Fig.1.Phase diagram for vesicles of DOPC/DPPC/Chol:(a) Phase diagram at 32 °C.Red dotted line denotes the composition whose Lo and Ld phases occupy the same surface area.Scale bar is 10 µm.(b) Size distribution of the domains.(c) Surface density of the domains.
图1(a)给出了降温5 min 的M1—M4 的GUVs显微图像.采用微畴面积、观测半球的微畴数量(>28 囊泡/组分)获取了微畴半径(r)及面密度,发现不同组分的相分离微畴尺寸、数量存在显著差异.M1—M3 的微畴半径(r)较大,面密度较低;而M4 的微畴尺寸较小,面密度较高.图1(b)和图1(c)给出了微畴尺寸与面密度的统计结果.M1—M3 微畴的平均半径分别是M4 的4.7,3.2,2.0 倍,但面密度是后者的10.1%,19.8%,30.0%.这表明前三个组分微畴更迅速地粗化.需要指出,在较高线张力条件下,微畴可出芽以降低其与大相的接触边长[8,16].但图1(a)中未观测到明显的出芽结构.低线张力(见下文)与低渗正膜张力抑制了膜曲率的产生.最终微畴完全融合形成两个完全分离的Lo-Ld大畴结构.
GUVs 微畴的时间演变(图2(a)和图2(b))表明微畴的融合次数具有组分依赖性.图2(c)对降温2.5—4.0 min 内单位膜面积内的融合次数(n)进行了统计.此时微畴的半径主要分布在0.4—10.5 µm 之间,能够准确地进行个体采集与分析.发现融合次数符合nM1>nM2>nM3>nM4,其中M1 在统计时段内的融合次数是M4 的19 倍,表明微畴融合的差异可能是造成不同组分微畴粗化差异的重要原因.而对融合与未融合微畴尺寸演变的统计(图2(d))表明,通过蒸发-凝聚方式的粗化是可以忽略的.这与之前报道的融合粗化主导了Lo-Ld相分离中微畴的粗化一致[7].线张力(λ)是微畴融合粗化的主要驱动力,反映了不同磷脂组分相溶性的差异[9,15].为考察实验中观测到的粗化差异是否由线张力产生,通过微畴的径向波动性分析了M1 —M4 的线张力(图3(a)).图3(b)给出了径向波动性谱线,图3(c)给出了对应傅里叶级数展开的波数和系数,通过(3)式对此数据进行线性拟合获得线张力(图3(d)).发现线张力符合λM1>λM4>λM2>λM3,所有组分的平均线张力都处于同一量级(0.1 pN).0.1 pN 量级被认为是微畴尺寸由纳米向微米转变的临界线张力[28].低于该量级,微畴受热扰动无法融合为显微镜可见的相分离.更为重要的是,线张力与粗化微畴尺寸(图1(b))的大小排序并不一致.Garcia-Saez 等[15]通过改变磷脂尾链长度产生了跨3 个量级的线张力,发现微畴的平均面积粗化率与线张力成正比.但在我们的研究中,由于所有组分的线张力均处于同一量级,其他因素可能在相畴融合粗化中发挥了重要作用.

图2 微畴的粗化 (a) M1 和(b) M4 的微畴融合粗化.部分典型微畴的扩散和融合被标记出来.其中绿色圆环表示发生融合,红色箭头表示相邻时刻间微畴的扩散方向.(c) 2.5—4.0 min 中M1—M4 单位膜面积的融合次数.(d) 微畴的尺寸演变.微畴Ⅱ,Ⅲ,Ⅳ融合生成微畴Ⅵ,产生了显著的微畴粗化.而未发生融合的微畴Ⅰ尺寸没有发生变化.标尺为10 µmFig.2.Coarsening of domains.Coarsening by coalescence of domains in (a) M1 and (b) M4.The diffusion and coalescence of some typical domains are marked.The green circles denote the coalescence.The red arrows denote the diffusion direction of domains between the adjacent images.(c) Number of coalescences in unit surface area between 2.5—4.0 min.(d) Time evolution of domain size.Coarsening of domains is produced by the coalescence of the domains Ⅱ,Ⅲ,Ⅳ to form domain Ⅵ.Size of the domain Ⅰ remains unchanged without coalescence.Scale bar is 10 µm.

图3 基于微畴径向波动性计算的线张力 (a) M1—M4 微畴的荧光显微图;(b) 微畴极角(θ)对应的径向波动性;(c) 由傅里叶级数展开的波数(k)和系数(ak,bk)计算的 与 1/(k2-1) 关系;(d) 磷脂组分依赖的线张力Fig.3.Line tension calculated by domain boundary fluctuation: (a) Fluorescence microscopy of domains in M1-M4;(b) radial fluctuation as a function of polar angle (θ);(c) relationship between and 1/(k2-1) calculated from the Fourier coefficients (ak,bk) and mode number (k);(d) dependence of line tension on lipid composition.
在微畴的融合粗化中,微畴需要经历扩散碰撞才能实现融合.进一步考察了微畴在膜内的扩散动力学.图4(a)和图4(b)给出了微畴的代表性运动轨迹.在考察的GUVs 中,M3 与M4 的微畴基本符合布朗运动,而M1 与M2 有大量微畴在部分时段不符合布朗运动.布朗运动微畴的均方位移(MSD)和时间呈线性关系(图4(a)蓝线,(5)式);而非布朗运动微畴的MSD 呈指数型增长(图4(a)红线).微畴的非布朗运动源于Liang 等[27]报道的融合促进的微畴扩散,即微畴融合时产生的磷脂层流场诱导了周边微畴的扩散.Yanagisawa 等[29]将其简化为微畴间吸引.图4(b)给出了一个融合促进微畴扩散的轨迹: 在周边微畴Ⅰ和Ⅱ融合前,微畴Ⅲ呈布朗运动(蓝色轨迹);微畴Ⅰ和Ⅱ融合后,微畴Ⅲ迅速向Ⅰ,Ⅱ方向迁移(红色轨迹).更多的融合促进的微畴运动见图2(a).需要指出,流场的动能是由微畴融合造成的线张力降低提供的.流场速率(v)为v~σ/η[27],其中σ=λ/h2~10-7N/m2(h为微畴与大相的厚度差)[15],η为大相黏滞系数.由v的表达式可得,降低大相黏滞性可显著提高流场速率.在M1 和M2 组分实验中,观测到较为显著的非布朗运动微畴,即强流场影响,表明M1 和M2的大相黏滞性可能比M3 和M4 低.
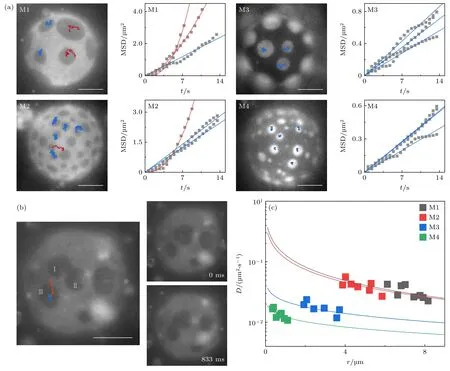
图4 微畴在膜内的扩散 (a) 典型的运动轨迹与MSD,红、蓝色分别标记了布朗运动与非布朗运动.(b) 融合促进的微畴扩散,在微畴Ⅰ和Ⅱ融合前,微畴Ⅲ呈布朗运动(蓝色轨迹);在微畴Ⅰ和Ⅱ融合后,微畴Ⅲ以近线性轨迹(红色)向Ⅰ,Ⅱ方向迁移.微畴Ⅲ在833 ms 内扩散迁移了约3 µm (右图).(c) 基于布朗运动的微畴统计获得的扩散速率-微畴尺寸关系.标尺为10 µmFig.4.Diffusion of domains in lipid membranes: (a) Typical trajectories and MSD.Brownian and non-Brownian motions are marked by blue and red colors.(b) Coalescence-induced domain diffusion.Before the coalescence of domains Ⅰ and Ⅱ,domain Ⅲunderwent Brownian motion (blue trajectory).After the coalescence,domain Ⅲ diffused to Ⅰ and Ⅱ through a nearly straight line(red trajectory).The diffusion distance of domain Ⅲ is around 3 µm in 833 ms (images at right).(c) Plot of diffusion coefficient versus domain size obtained from the Brownian motion of the domains.Scale bar is 10 µm.
基于符合布朗运动微畴的均方位移,估算了影响微畴扩散的重要参量η.首先,采用(5)式计算布朗运动微畴的侧向扩散速率D(图4(c)).发现对于相近尺寸的微畴,在Ld大相的扩散速率高于Lo大相.这源于不同相态磷脂膜自由体积含量的差异[30].磷脂层中侧向扩散的本质是磷脂分子不断占据可用自由体积的过程[31].Lo相磷脂排布相对有序,自由体积含量较低,因此其中内含物的扩散较慢.与之相对,Ld相磷脂排布的相对无序产生了较快的侧向扩散.随后,基于Petrov 和Schwille 等[32]构建的Hughes-Pailthorpe-White (HPW)经验关系式数值求解大相的黏滞系数:

其中ε=2rηm/η,ηm为溶液黏滞系数;γ为欧拉常数;c1,c2,b1,b2为常数.获得的η值如图5(a)所示.可以看出,Lo大相的黏滞系数在10—8—10—7Pa·s·m,Ld大相的黏滞系数在10—9Pa·s·m (图5(a)),这与表面剪切流变实验报道的磷脂膜黏滞性范围是一致的[33].并且黏滞系数符合ηM4>ηM3>ηM2>ηM1(图5(a)).M4 的黏滞系数比M1 高两个量级.图5(b)给出了大相黏滞系数与对应微畴尺寸(降温5 min)的关系,两者的同向变化说明了两者的内在相关性.下文将半定量地讨论大相黏滞性与微畴粗化的关系.
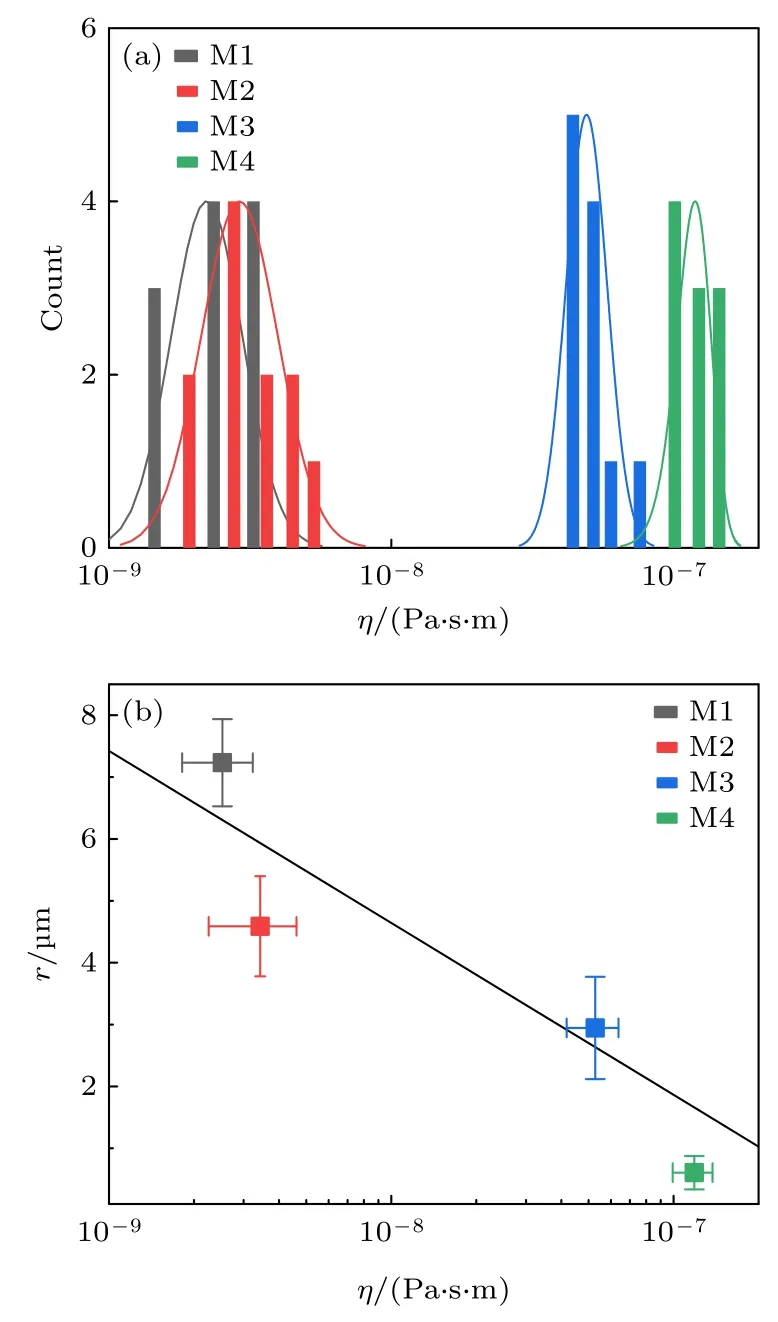
图5 大相的黏滞性 (a) 数值求解的大相黏滞系数;(b) 大相黏滞系数与降温5.0 min 的M1 —M4 的微畴尺寸Fig.5.Bulk viscosity: (a) Numerically computed bulk viscosity;(b) plot of the bulk viscosity versus domain size in M1-M4 (5.0 min after the temperature quench).
首先讨论产生ηM4>ηM3>ηM2>ηM1的原因.胆固醇可能显著影响了大相磷脂的密排结构,从而改变了大相的黏滞性[31,34].通过DPH 荧光光谱实验考察了胆固醇含量对膜黏滞性的影响.DPH 的摆动影响了发射光各向异性,反映着周边磷脂双层疏水内核的黏滞性[26].如图6(a)所示,单组分DOPC和DPPC 膜分别具有DPH 各向异性(a)的最低值与最高值,对应着它们所处的液相(Lα)、凝胶相(Lβ)的极低与极高黏滞性(~10-10Pa·s·m[34],~10-6Pa·s·m[35]).Chol 的加入引起了a的变化.对于DOPC/Chol 膜,a随着Chol 含量的提高而增大,表明Chol 使得Lα相磷脂膜的黏滞性提高.而对于DPPC/Chol 膜,Chol 含量的提高降低了a,表明Chol 降低了Lβ相磷脂膜的黏滞性.基于以上结果,给出Chol 影响多组分GUVs 中大相黏滞性的机制示意图(图6(b)).当Lo大相中的Chol与DPPC 含量之比提高时(Chol/DPPCM4=0.29,Chol/DPPCM3=0.97),性质更偏离DPPC 的Lβ相,导致ηM3<ηM4.当Ld大相中的Chol 与DOPC含量之比提高时(Chol/DOPCM1=0.17,Chol/DOPCM2=0.27),膜性质更偏离DOPC 的Lα相,导致ηM2>ηM1.以上结果表明,可通过改变胆固醇的相对含量调节大相黏滞性,从而间接影响磷脂膜的相分离动力学.

图6 Chol 影响的磷脂膜黏滞性 (a) 掺杂DPH 的DPPC/Chol 与 DOPC/Chol 组分GUVs 的荧光光谱与各向异性(32 °C);(b) 黏滞性变化对应的分子排布示意图.磷脂排布有序度的提高造成膜黏滞性提高[34]Fig.6.Membrane viscosity influenced by Chol: (a) Fluorescence spectrum and anisotropy of DPH-doped DPPC/Chol and DOPC/Chol GUVs (32 °C);(b) schematic illustration for the molecular packing associated with the different membrane viscosity.The increase of the packing order increases the viscosity[34].
进一步考察大相黏滞性与微畴粗化率间的关系.实验中降温迅速完成(1.0 min),以消减二次微畴成核造成的尺寸波动.图7 在对数坐标系中给出了归一化的微畴的平均半径与时间关系.发现所有组分的微畴均满足r=tα.但大相黏滞性低实验拟合的α值 (αM1=0.96,αM2=0.93)显著高于大相黏滞性高实验的拟合值 (αM3=0.52,αM4=0.50).相近地,Liang 等[27]在微畴融合诱导的扩散体系中观测到α~1,Tayebi 等[36]在高黏滞的吸附磷脂多层体系中观测到α~0.5 .为理解大相黏滞性相关的微畴粗化标度律,假设大相中均匀分布着许多尺寸相同的微畴.当微畴尺寸较小时,微畴面密度较高导致它们间距较低;但当微畴融合粗化至较大尺寸时,微畴密度降低导致它们间距提高.因此,微畴的半径r(t)近似与微畴的间距l(t) 成正比.当大相黏滞性较高时,微畴需要以布朗运动扩散l(t)的距离与周边微畴接触融合,即l(t)2∝Dt.由于r(t)∝l(t),因此r(t)2∝Dt.代入HPW 的Saffman-Delbrück 简化式[25]:D=(需满足η>ηmr,实验条件下的估算值见表2),忽略对数项近似得到:
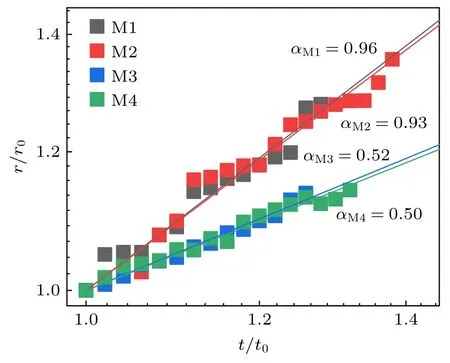
图7 大相黏滞性依赖的微畴尺寸与时间标度律.r 与t被归一化以方便比较(下标0 代表初始时刻)Fig.7.Bulk viscosity-depended scaling relation between domain size and time.r and t are normalized for comparison of the data (the subscript 0 denotes the initial).

(8)式与高黏滞的M3 和M4 实验结果相近.而当大相黏滞性较低时,微畴融合引起的流场可促进周边微畴的靠拢.微畴间融合的时间tf近似为tf∝l(t)/v.微畴尺寸的增长满足 dr(t)/dt=.而由于r(t)∝l(t),可得

(9)式与低黏滞的M1 和M2 实验结果相近.因此,高低黏滞性下不同的微畴尺寸-时间标度律是由不同的微畴扩散机制引起的.需要指出,以上α值与早些无膜张力体系中观测到的α≤1/3 具有区别.较小的α值可能源于膜形变等因素[37,38].低渗正膜张力实验条件抑制了膜的形变与热波动,从而促进了相分离的进行[19].综上所述,在线张力较低的多组分磷脂相分离中,大相黏滞性可在微畴的粗化动力学中发挥关键作用.
深入理解GUVs 微畴的粗化机制对深入理解脂质筏动力学至关重要.我们的研究给出了大相黏滞性引起的微畴扩散与粗化机制.但研究尚未给出高/低黏滞性的准确分界值.ηmr可能是重要的参考量(表2).当η<ηmr,周围水媒介与磷脂层的动量交换是可忽略的.而当η>ηmr,微畴融合产生的流场可能将动能显著地耗散于周边水媒介,从而降低其对微畴扩散的影响.之后的研究将对该分界值进行深入探索.

表2 实验条件下 η 与 ηmr 的估算值(降温后2.5—4.0 min)Table 2.Estimated values of η and ηmr at the experiments (2.5-4.0 min after the temperature quench).
4 结论
本文通过单微畴跟踪、径向波动性分析等手段定量地研究了微畴的粗化动力学.发现当大相黏滞性较低,微畴融合产生的流场促进了微畴扩散,加速了微畴融合粗化;当大相黏滞性较高,微畴通过布朗运动扩散,融合粗化较慢.在两种情况下,分别满足r(t)∝t0.5与r(t)∝t1的微畴粗化标度律.此外,大相黏滞性可由胆固醇的相对含量调节,变化的方向具有大相种类依赖性.研究深化了多组分磷脂相分离机制的理解,为考察细胞脂质筏的动态行为、调控膜表面的分子组装提供了参考.
感谢南京大学物理学院向雅心博士关于理论建模的讨论.
