热休克蛋白A12B介导COX-2表达调控心肌缺血再灌注损伤中心肌细胞线粒体自噬的作用研究
2022-09-27王书君谭慧琼
王书君,谭慧琼,袁 杰
(1.海南医学院第一附属医院老年医学科,海南 海口 570102;2.中国医学科学院阜外医院心血管内科,北京 100037;3.新疆维吾尔自治区人民医院心内科,新疆 乌鲁木齐 830001)
急性心肌梗死是全球范围内常见的心血管疾病,及时开通梗死相关动脉,恢复缺血心肌的供血,是挽救垂死心肌及防止梗死范围再次扩大的关键。但有研究发现,心肌缺血一段时间后恢复血流灌注会加重心肌结构和功能的损伤,导致心功能下降和恶性心律失常,这一过程称为心肌缺血再灌注(myocardial ischemia-reperfusion,MI/R)损伤[1-2]。目前,MI/R损伤仍是治疗心肌缺血所面临的主要难题。因此,如何在早期恢复冠脉血流的基础上减少甚至消除MI/R损伤的发生具有重要的临床意义。
热休克蛋白A12B(heat shock protein A12B,HSPA12B)于2003年从动脉粥样硬化病变中克隆出来,归类为热休克蛋白70(HSP70)家族的一员,包含非典型的ATPase结构域[3]。与HSP70家族其他成员的普遍性表达不同,HSPA12B在内皮细胞中特异性表达,能够抵御多种有害因素。已有研究表明,HSPA12B能够在心肌梗死和脑缺血后发挥对组织器官的保护作用[4-5],还能够改善脓毒症引起的心脏功能障碍[6],但相关的作用机制尚未明确。
为进一步确定HSPA12B在MI/R损伤中的具体作用及机制,本研究通过构建MI/R大鼠模型并给予含HSPA12B的慢病毒液预先处理,观察HSPA12B对MI/R心肌组织损伤及心肌组织内环氧化酶-2(cyclooxygenase-2,COX-2)表达的影响,并检测线粒体自噬途径的变化情况,旨在揭示HSPA12B调控MI/R损伤的作用机制,为MI/R损伤的治疗提供参考。
1 材料与方法
1.1 实验动物
SPF级健康SD大鼠,雄性,4周龄,体质量(200±20)g,购自海南药物研究所有限责任公司,饲养于无特定病原体屏障环境中,温度(23±2)℃,相对湿度(50±5)%,12 h/12 h明暗周期交替,实验期间自由饮水、摄食。本实验获得中国科学院阜外医院动物伦理委员会审核批准(K20211021208)。
1.2 主要试剂
空白慢病毒载体pLVX-NC和含HSPA12B慢病毒载体pLVX-HSPA12B的合成和包装均由上海吉玛制药技术有限公司完成。血清肌酸激酶同工酶(creatine kinase isoenzymes,CK-MB)和乳酸脱氢酶(lactate dehydrogenase,LDH)特异性ELISA试剂盒购自上海慧颖生物公司,HE染色试剂盒购自北京索莱宝生物公司,TUNEL细胞凋亡检测试剂盒购自上海翌圣生物公司,免疫组织化学染色试剂盒购自美国Thermo Fisher公司,DAB显色液、RIPA裂解液、考马斯亮蓝试剂盒和ECL发光试剂液购自上海碧云天生物研究所,兔抗人COX-2多克隆、兔抗人微管相关蛋白1轻链3β(microtubule-associated protein 1 light chain 3 beta,LC3β)多克隆、兔抗人Parkin多克隆、兔抗人PINK1多克隆及兔抗鼠GAPDH多克隆等抗体均购自英国Abcam公司。其他试剂均为国产分析纯。
1.3 分组与处理
将40只SD大鼠按照随机数字表法分为4组,每组10只。假手术组大鼠仅暴露心脏,不进行MI/R处理;模型组大鼠心肌内注射0.9%氯化钠注射液20 μL,7 d后构建MI/R模型;pLVX-NC组大鼠心肌内注射20 μL pLVX-NC慢病毒液,7 d后构建MI/R模型;pLVX-HSPA12B组大鼠心肌内注射20 μL pLVX-HSPA12B慢病毒液,7 d后构建MI/R模型。注射操作如下:将大鼠消毒后麻醉,仰卧位固定,气管插管并连接动物呼吸器进行机械通气;在大鼠左胸第4和第5肋骨之间剪开皮肤,使心脏完全暴露,采用微量注射器分4点在各组大鼠心肌中注射相应液体,留置注射针2 min,闭合胸腔,肌内注射1.5 mL/kg硫酸庆大霉素抗感染,每日1次,持续3 d。
1.4 动物造模
参考文献[7]方法构建MI/R大鼠模型。将大鼠消毒后,腹腔注射40 mg/kg戊巴比妥钠麻醉,然后固定在动物实验台上,气管插管并连接动物呼吸器进行机械通气。在大鼠左胸第4和第5肋骨间剪开皮肤,使心脏完全暴露,用丝线在冠状动脉左前降支下约2 mm处进行结扎,以阻塞左冠状动脉前降支,心电图观察ST段抬高0.1 mV左右,持续结扎40 min进行缺血处理,之后释放结扎丝线,再灌注120 min,待ST段下降到缺血时的0.05 mV,即成功构建大鼠MI/R模型。随后立即闭合胸腔,缝合切口,消毒,肌内注射1.5 mL/kg硫酸庆大霉素抗感染,每日1次,持续3 d。
1.5 ELISA检测心肌损伤指标
处理结束后,各组大鼠经尾静脉采血2 mL,室温静置2 h,4 ℃低温以4 000 r/min离心20 min,制备血清。将样品加入各反应孔,使用大鼠特异性ELISA试剂盒测定各组大鼠血清CK-MB和LDH含量。
1.6 HE染色观察心肌组织病理学改变
采血后处死各组大鼠,无菌环境下解剖取各组大鼠心肌组织,清洗干净,在4%多聚甲醛中固定,然后经梯度酒精脱水,二甲苯透明,常规石蜡包埋,制备厚度约为5 μm的切片。将切片脱蜡水化,苏木素染色10 min,流水冲洗多余染液,1%酒精—盐酸分化,水洗,反蓝,伊红染色1 min,再次经梯度酒精脱水,二甲苯透明,随后中性树胶封固,光学显微镜下观察各组大鼠心肌组织病理学变化,并摄取图片。
1.7 TUNEL染色检测细胞凋亡情况
取制备的各组大鼠心肌组织切片,以梯度酒精脱水,二甲苯透明,PBS清洗,加入20 μg蛋白酶K溶液,室温水解15 min,蒸馏水清洗;加入含2%过氧化氢的PBS,室温反应5 min,PBS清洗,弃掉组织周围多余染液,滴加TdT试剂液,置于湿盒中37 ℃孵育60 min;终止反应后,PBS清洗,使用新鲜配制的DAB溶液室温显色5 min,脱水透明,随后中性树胶封固,光学显微镜下随机选择5个200倍镜视野进行观察。阳性细胞呈褐色,计数视野下200个细胞与其中的阳性细胞数,计算各组大鼠心肌组织内TUNEL染色阳性细胞率。
1.8 免疫组织化学染色检测心肌组织中COX-2、LC3β表达
各组大鼠心肌组织切片以二甲苯脱蜡,梯度酒精脱水,柠檬酸钠修复抗原,0.3%过氧化氢液室温孵育10 min,PBS浸泡清洗,再以10%山羊血清进行封闭。结束后,分别滴加兔抗人COX-2(1∶100)、LC3β(1∶200)多克隆抗体,4 ℃孵育过夜。次日,弃掉原液,PBS清洗,滴加对应的二抗(1∶5 000),室温孵育1 h后采用DAB溶液室温显色,冲洗干净,随后苏木精复染,脱水透明,中性树胶封固,光学显微镜观察组织内着色情况。阳性染色为胞质或胞核呈棕色至深褐色,随机选择5个视野,利用Image J软件进行分析,计算各组大鼠心肌组织内COX-2、LC3β阳性表达率。
1.9 Western blot检测相关蛋白的表达
将各组大鼠心肌组织在无菌环境下剪碎,加入液氮研磨,添加适量RIPA裂解液,置于冰上裂解,以提取组织总蛋白,考马斯亮蓝法测定。取各组蛋白样品,98 ℃水浴加热,并制备10%SDS-PAGE,取等量的蛋白样品进行上样后,经电泳分离蛋白,80 V恒压电泳20 min,110 V再电泳60 min,恒压100 V处理90 min,将分离的蛋白转至PVDF膜。5%脱脂奶粉室温封闭1 h,加入相应一抗工作液,4 ℃孵育过夜。次日,弃掉原液,TBST洗膜后加入相应的二抗,室温孵育2 h,TBST再次洗膜,ECL显色,凝胶成像系统拍照,以GAPDH作为内参蛋白,Image J软件分析蛋白条带灰度值,计算LC3Ⅰ、LC3Ⅱ、Parkin及PINK1蛋白的相对表达水平。
1.10 统计学分析
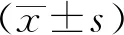
2 结果
2.1 各组大鼠血清心肌损伤标志物水平
与假手术组比较,模型组大鼠血清CK-MB和LDH的含量均明显升高(P<0.05);与模型组和pLVX-NC组比较,pLVX-HSPA12B组大鼠血清CK-MB和LDH的含量均明显降低(P<0.05),见表1。

表1 各组大鼠血清心肌损伤指标水平比较
2.2 各组大鼠心肌组织病理变化
假手术组大鼠心肌组织结构正常,组织内细胞排列较为整齐,心肌纤维紧密,无明显的肿胀及紊乱现象;模型组和pLVX-NC组大鼠心肌纤维肿胀变大,出现断裂,并伴大量炎性细胞浸润;与模型组和pLVX-NC组比较,pLVX-HSPA12B组大鼠心肌组织病理损伤现象得到缓解,心肌纤维断裂现象减少,心肌细胞排列较为整齐(图1)。
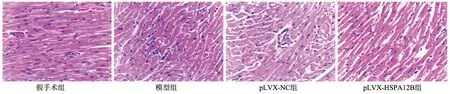
图1 各组大鼠心肌组织病理学形态(HE染色×100)
2.3 各组大鼠心肌组织细胞凋亡情况
TUNEL染色结果显示,模型组大鼠心肌组织内TUNEL阳性细胞率较假手术组明显升高(P<0.05),表明凋亡细胞增加;pLVX-HSPA12B组大鼠心肌组织内TUNEL阳性细胞率较模型组和pLVX-NC组明显下降(P<0.05),表明凋亡细胞减少(图2)。
a:TUNEL染色(×200);b:TUNEL染色阳性细胞率 *:与假手术组比较,P<0.05;#:与模型组比较,P<0.05;△:与pLVX-NC组比较,P<0.05
2.4 各组大鼠心肌组织COX-2表达
与假手术组比较,模型组大鼠心肌组织内染色加深,COX-2阳性表达率明显升高(P<0.05);与模型组和pLVX-NC组比较,pLVX-HSPA12B组大鼠心肌组织内染色明显变浅,COX-2阳性表达率明显降低(P<0.05),见图3。

a:免疫组织化学染色(×100);b:COX-2阳性表达率 *:与假手术组比较,P<0.05;#:与模型组比较,P<0.05;△:与pLVX-NC组比较,P<0.05
2.5 各组大鼠心肌细胞线粒体自噬水平
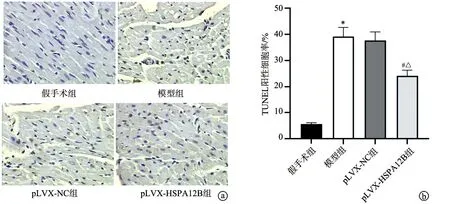
与假手术组比较,模型组大鼠心肌组织内染色较少,且颜色变浅,LC3β阳性表达率明显下降(P<0.05);而相较于模型组和pLVX-NC组,pLVX-HSPA12B组大鼠心肌组织内染色加深,LC3β阳性表达率明显上升(P<0.05),见图4。

a:免疫组织化学染色(×100);b:LC3Ⅱ/LC3Ⅰ比值;c:Parkin蛋白相对表达;d:PINK1蛋白相对表达 *:与假手术组比较,P<0.05;#:与模型组比较,P<0.05;△:与pLVX-NC组比较,P<0.05
2.6 各组大鼠心肌组织自噬相关蛋白表达
与假手术组比较,模型组大鼠心肌组织内LC3Ⅱ/LC3Ⅰ比值下降(P<0.05),Parkin、PINK1蛋白的表达水平明显下调(P<0.05);与模型组和pLVX-NC组比较,pLVX-HSPA12B组大鼠心肌组织内LC3Ⅱ/LC3Ⅰ比值上升(P<0.05),Parkin、PINK1蛋白的表达水平明显上调(P<0.05),见图5。
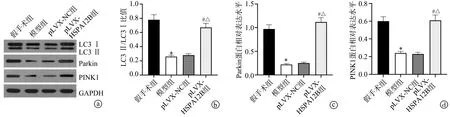
a:各蛋白条带;b:LC3Ⅱ/LC3Ⅰ比值;c:Parkin蛋白相对表达水平;d:PINK1蛋白相对表达水平 *:与假手术组比较,P<0.05;#:与模型组比较,P<0.05;△:与pLVX-NC组比较,P<0.05
3 讨论
MI/R诱导的心肌细胞死亡是导致冠心病患者死亡的主要病理因素之一。MI/R会引发一系列复杂的炎症反应及严重的组织损伤和心律失常,阻止心脏收缩功能恢复,并导致缺血组织中细胞死亡[2,8]。目前,MI/R引发的一系列复杂性损伤已成为全球范围内致残和致死的主要因素,也是心血管疾病患者死亡的主要原因[9]。而且没有切实可行的解决方案能够彻底避免MI/R损伤,迫切需要制定出针对治疗MI/R损伤的新策略。因此,本研究通过使用含HSPA12B的慢病毒液预先处理大鼠后构建MI/R模型,了解其对MI/R损伤的潜在作用机制,以期为MI/R的治疗策略提供参考。
HSPA12B作为内皮细胞中特异性表达的热休克蛋白,参与内皮细胞的增殖和迁移,能够促进血管生成,现已证明HSPA12B在一些疾病中发挥重要的治疗作用。Zhao等[10]研究表明,HSPA12B过表达可通过eNOS依赖性机制促进缺血性中风慢性期小鼠的神经功能恢复并提高其存活率;Zhang等[6]研究发现,HSPA12B可通过调节靶向黏附分子的miR-126表达来预防败血症诱导的严重心肌病,从而减少心肌中免疫细胞的积累;Thirunavukkarasu等[11]的研究指出,HSPA12B基因疗法可以改善后肢缺血小鼠的运动功能,促进新生血管形成,并减少组织纤维化。本研究结果显示,与模型组和pLVX-NC组比较,在经HSPA12B预处理的MI/R大鼠模型中,血清中损伤标志物CK-MB和LDH的含量均下降,心肌组织损伤现象明显减轻,进一步说明了HSPA12B对MI/R损伤具有良好的缓解作用。
COX是前列腺素生物合成的限速酶,其有COX-1和COX-2两种异构体。COX-1在许多组织中表达,主要在组织稳态中发挥作用;相比之下,COX-2作为一种诱导性酶,是各种炎症过程的中心环节,负责在炎症和伤口愈合部位产生前列腺素[12]。COX-2在炎症血管中表达上调,并在动脉粥样硬化、动脉瘤和动脉球囊损伤中对病程发展起到促进作用[13]。另外,Yin等[14]通过将主动脉血管内皮细胞暴露在PM2.5环境中后发现,COX-2的表达水平明显上调,而在暴露于PM2.5环境前用特定COX-2抑制剂处理血管内皮细胞后,不仅完全阻止了细胞凋亡,而且还减轻了炎症反应。越来越多的研究表明,COX-2与心肌梗死疾病的进展有关,Abbate等[15]研究表明,COX-2在心肌梗死患者的心肌细胞中高表达,并与细胞凋亡呈正相关;贾丹等[16]指出,抑制COX-2活性能够降低体外缺氧/复氧实验模拟的MI/R中促炎因子的转录,并对H9C2心肌细胞具有保护作用。鉴于此,本研究通过检测各组MI/R大鼠心肌组织内COX-2表达发现,与模型组和pLVX-NC组比较,经HSPA12B预处理的MI/R大鼠心肌组织内COX-2阳性表达率明显降低。由此推测,HSPA12B可能通过介导COX-2在MI/R损伤中发挥作用。
终末分化心肌细胞的死亡是各种心脏疾病发生的主要原因,包括心力衰竭、心肌梗死及MI/R损伤。在心脏病发病过程中,细胞凋亡和坏死都能够导致心肌细胞死亡,而心肌细胞死亡是MI/R损伤中心脏功能衰竭的核心特征[17]。心肌细胞死亡是不可逆的,并伴细胞膜电位的快速丧失,从而导致细胞肿胀、破裂、溶解及后续炎症的发生。线粒体自噬指细胞降解自身线粒体的过程,该途径可以清除MI/R损伤下心脏中功能失调的线粒体,以减轻病理损伤[18]。研究表明,心肌细胞中线粒体自噬的机制是由胞质E3泛素连接酶Parkin和线粒体膜激酶PTEN诱导的推定激酶PINK1介导,PINK1在线粒体中选择性稳定,其内膜上的电位降低,并将Parkin募集到线粒体以激活其E3泛素连接酶活性,导致线粒体自噬并通过自噬机制去除受损的线粒体[19]。此外,Parkin非依赖性线粒体自噬途径由Bcl-2、BNIP3或BNIP3L介导,这些受体激活后可直接与LC3β结合,促进缺氧条件下的线粒体自噬发生。本研究经检测发现,与模型组和pLVX-NC组比较,经HSPA12B预处理的MI/R大鼠模型心肌组织内LC3β阳性表达率上升,Parkin、PINK1蛋白的表达水平明显上调,LC3Ⅱ/LC3Ⅰ比值也升高。由此推测,HSPA12B作用下增加了MI/R大鼠心肌细胞线粒体自噬,从而发挥对心脏组织的保护作用。
综上,大鼠MI/R损伤会导致COX-2表达升高,线粒体自噬功能降低,心肌组织损伤,而HSPA12B能够抑制COX-2表达,并增加线粒体自噬,从而逆转MI/R损伤。本研究从线粒体自噬入手,为探究以HSPA12B为靶点改善MI/R损伤提供了实验依据。
