Mn(Ⅱ)在伊利石(001)面和(010)面吸附的密度泛函研究
2022-09-20王庆鹤蔡深文李雅慧曾伯平
杜 佳, 王庆鹤, 蔡深文, 樊 海, 李雅慧, 陈 军, 曾伯平
(1. 遵义师范学院资源与环境学院, 遵义 563006; 2. 安徽理工大学材料科学与工程学院, 淮南 232001; 3. 遵义师范学院生物与农业科技学院, 遵义 563006)
重金属对水体和土壤的污染一直是环境治理的难题,黏土矿物对环境中重金属的吸附主要集中在:(1)为开发黏土矿物材料,对矿物表面进行改性以提高吸附性能[1];(2)通过一定的物理化学方法促使重金属离子从黏土矿物表面解吸,实现土壤的净化或黏土吸附材料的回收利用[2-3]。研究内容主要探究宏观规律,然而对于更深层次的吸附机理的探讨较为匮乏。随着计算化学的发展,密度泛函理论被广泛应用于吸附微观机理的研究。郑刘春等[4]通过密度泛函理论多角度的阐述了螯合纤维对水体中Cd(Ⅱ)的吸附机理。CHEN等[5]通过DFT研究Hg(Ⅱ)在高岭石(001)面的吸附,结果表明Hg(Ⅱ)与表面O形成共价键,且主要是Hg 5d与O 2p轨道间的相互作用而成键。SANCHEZ等[6]借助量子力学理论研究金属Fe、Cu和Zn在伊利石的分布,结果表明离子的高化合价态更容易与伊利石晶体结合,形成稳定结构。可见,密度泛函理论能深入原子层面,为吸附微观机理的研究提供了可行的手段。伊利石作为一种黏土矿物,在地壳中广泛分布。为探究Mn(Ⅱ)在伊利石表面的吸附微观机理。本文以伊利石的(010)面和(001)面为研究对象,采用密度泛函理论研究Mn(Ⅱ)在伊利石表面的吸附行为,通过对吸附活性位、吸附能、吸附构型、态密度和电荷进行分析,以揭示吸附机理,研究结果可为吸附新材料的开发及土壤污染的治理提供理论基础。
1 研究方法
1.1 表面模型建模
伊利石晶胞采用DRITS模型[7],为满足伊利石的结构要求,构建2×1×1的超晶胞,并设置晶格取代,最终晶胞为K0.5Al2(Si4Al0.5)O10(OH)2。为研究Mn(Ⅱ)在伊利石不同晶面的微观吸附机理,构建(001)面和(010)面的表面模型,并在表面上方添加1.5 nm厚的真空层,以避免周期性的影响。对表面进行结构优化,优化后的伊利石表面模型见图1,在图1A伊利石(001)面中,表面结构由上下2层SiO4四面体以及中间AlO6八面体组成,即TOT结构。(001)面的Si—O环内存在Al对Si的晶格取代而使表面荷负电,因而以K+作为平衡离子、平衡电荷;在图1B的(010)面中,裸露的Si和Al在环境中形成羟基基团,如≡Si—OH、≡Al—OH[8]。通过模型的构建,形成82个原子的(001)面和拥有94个原子的(010)面。
1.2 结构优化
采用基于第一性原理的Dmol3模块[9]对伊利石超晶胞进行结构优化,并选用广义梯度(GGA)的PBE交换相关泛函[10],设置Grimme色散校正。采用全电子体系计算,选择双数值正极化(DNP)原子中心基组,运用BFGS 优化算法,将自洽场收敛精度设定为1×10-6eV/atom。几何优化的收敛标准为:原子的最大位移为5×10-4nm,原子间的最大作用力为2×10-2Ha/nm,体系总能量的变化为1×10-5Ha,所有计算均在倒易空间中进行,布里渊区[11]k点采样选择1×2×1。表1是伊利石的晶格常数,模拟值与DRITS模型的实验值基本一致,表明密度泛函理论计算的参数设置具有合理性。

表1 伊利石晶格常数
表面模型及吸附体系优化采用与体相一致的交换关联函数和收敛标准。为避免吸附体系在真空环境下运行,选择COSMO模式,吸附环境在水介质中,介电常数为78.54,且通过晶格取代或增减平衡离子实现吸附体系的荷电中性。k点限制在г点。Mn(Ⅱ)在1.5 nm×1.5 nm ×1.5 nm的空间,k点采样限制在г点,优化标准与表面模型一致。
1.3 吸附能计算
通过吸附能的计算分析Mn(Ⅱ)在表面不同吸附位的强弱,其在伊利石表面的吸附能计算公式:
Eads=E[Mn—illite]-E[illite]-E[Mn],
(1)
其中,E[Mn-illite]表示Mn(Ⅱ)在表面吸附后体系的总能量;E[illite]为伊利石表面模型的能量;E[Mn]为Mn(Ⅱ)的能量;Eads为Mn(Ⅱ)在表面的吸附能,其中正、负分别表示吸热反应和放热反应,在放热反应中,绝对值越大表示吸附越稳定;在吸热反应中,吸附不能自发进行,吸附不稳定。
2 结果与讨论
2.1 吸附点分布
伊利石在(001)面的硅氧环有Al取代,能吸附平衡离子,静电作用较强,分析(001)面静电势的分布规律,能查清吸附活性位。图2为Mn(Ⅱ)与(001)面的静电势和初始吸附位分布,颜色的差异代表静电势的变化,由蓝色→白色→红色,表示静电势由负电性到正电性的转变。图2A为(001)面的静电势分布,在硅氧环的氧原子被蓝色包裹,存在负电性,且在环中央有蓝色区域的重叠,存在较强的负电性。这与蒙脱石在(001)面的静电势基本相似,在硅氧环内存在较强的负电势[12]。图2B为Mn(Ⅱ)的静电势,Mn(Ⅱ)被红色包裹,存在较强的正电势。根据电势的分布特征,在(001)面设置了不同Mn(Ⅱ)的吸附位,图2C为Mn(Ⅱ)初始吸附位的分布,包括:空穴位H1~H3、表面氧原子顶位T1~T3、氧原子之间或氧与硅氧环空穴位间的桥位B1~B4。将Mn(Ⅱ)置于以上氧原子空穴位、顶位、桥位处进行模拟计算,以获得吸附稳定构型。
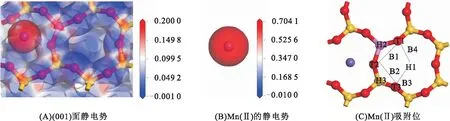
图2 Mn(Ⅱ)与(001)面的静电势和初始吸附位分布
伊利石(010)面有羟基基团暴露,易与重金属离子发生鳌合反应[13]。前线轨道理论表明最高占据轨道(HOMO)和最低未占据轨道(LUMO)是决定体系化学反应的关键[14]。因而,通过前线轨道分析能获得吸附位。图3为Mn(Ⅱ)与(010)面的前线轨道和初始吸附位分布,绿色为HOMO轨道,紫色为LUMO轨道。图3A为(010)面的前线轨道分布,羟基的氧原子被绿色包裹,主要为HOMO轨道,图3B为Mn(Ⅱ)的前线轨道分布,Mn(Ⅱ)被紫色包裹,主要为LUMO轨道。根据前线轨道理论,HOMO轨道与LUMO轨道的结合为最佳吸附组合,因此,对(010)面的吸附位进行设置。图3C为(010)面的Mn(Ⅱ)初始吸附位,其中羟基间的空穴位有H1~H3,羟基氧顶位T1~T5,羟基氧桥位B1~B7。模拟过程中将Mn(Ⅱ)布置在吸附位并进行结构优化,计算吸附能,以确定不同的稳定构型。
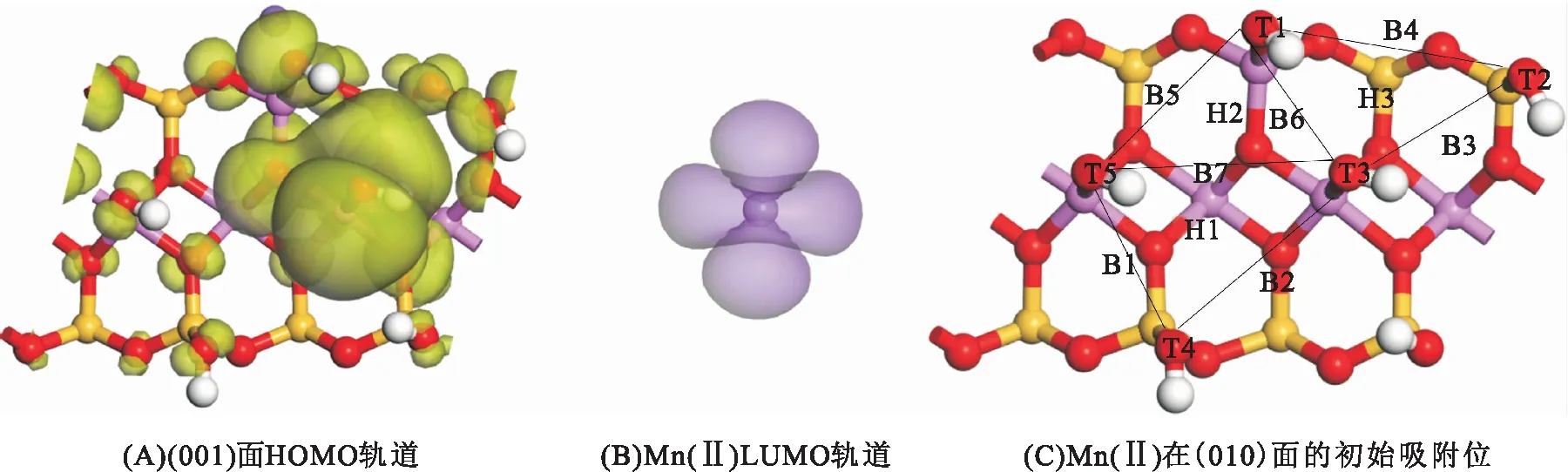
图3 Mn(Ⅱ)与(010)面的前线轨道和初始吸附位分布
2.2 吸附构型分析
通过在初始吸附位进行吸附体系的模拟计算,获得吸附能及吸附构型。图4为Mn(Ⅱ)在伊利石表面的吸附能,吸附能均为负,表明Mn(Ⅱ)在表面吸附为放热反应,吸附可以自发进行,吸附能绝对值越大,吸附越稳定。
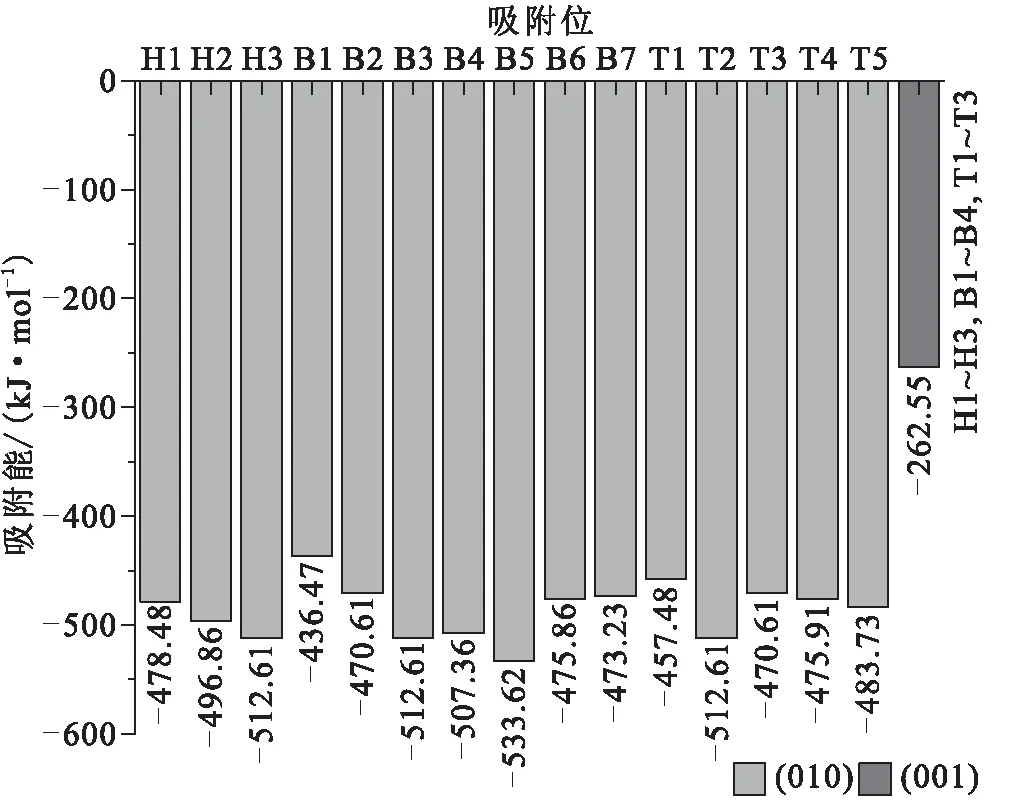
图4 Mn(Ⅱ)在伊利石表面的吸附能
在伊利石(001)面的吸附能均为-262.55 kJ/mol,其绝对值低于在(010)面的吸附能(-436.47~-533.62 kJ/mol)的绝对值,结合图5中Mn(Ⅱ)在伊利石表面的吸附构型进行进一步分析。图5A中 (001)-A为在(001)面的吸附构型,虽然有不同初始吸附位,但是吸附的稳定构型只有一种,位于伊利石(001)面的硅氧环内,且与活性氧原子存在共价键。在伊利石(010)面的吸附能随着初始吸附位的不同而不同。具体表现为:(1)在图5的D、E、F、K和M构型中,即(010)-B3、(010)-B4、(010)-B5、(010)-H3、(010)-T2吸附体系,Mn(Ⅱ)的吸附位于≡Al—OH或≡Si—OH构成的3羟基基团的空穴处,与羟基中的氧原子形成3条共价键,吸附能在-507.36~-533.62 kJ/mol范围,其中3个羟基为≡Al—OH时,基团空穴处的Mn(Ⅱ)吸附能最大,即(010)-B5构型;(2)在图5的I、J、O和P构型中,即(010)-H1、(010)-H2、(010)-T4和(010)-T5吸附体系,Mn(Ⅱ)吸附于≡Al—OH或≡Si—OH构成的2个羟基基团的氧桥位处,且形成2条共价键,吸附能在-475.91~-496.86 kJ/mol范围,其中吸附点位于2个≡Si—OH氧桥位的吸附能最低,即(010)-T4构型;(3)在图5的B、C、G、H、L和N构型中,即(010)-B1、(010)-B2、(010)-B6、(010)-B7、(010)-T1和(010)-T3吸附体系,Mn(Ⅱ)吸附于≡Al—OH或≡Si—OH单个羟基基团上方,且形成1个共价键,吸附能为-436.47~-475.86 kJ/mol,且在吸附位于≡Si—OH上方的吸附能最低,即(010)-B1。综上所述,在伊利石(001)面的吸附能小于在伊利石(010)面的吸附能,(010)面的吸附能随着共价键数目的增多而增大,≡Si—OH对Mn(Ⅱ)的吸附能小于≡Al—OH。

图5 Mn(Ⅱ)在伊利石表面的吸附构型
为进一步探究吸附构型,选取不同吸附面的最稳定构型进行深入分析。图6为Mn(Ⅱ)在伊利石表面吸附的最稳定构型,表2为吸附稳定构型参数,构型结合参数进行分析。图6A为(001)-A构型,Mn(Ⅱ)吸附于空穴中央,与OS1形成键长为0.218 9 nm的共价键,这是由于荷电的(001)面产生极化诱导,使得活性OS1与Mn(Ⅱ)形成共价键。LI等[15]的研究说明这种共价键的强度受溶液介质的pH、电解质类型及浓度的影响。Mn(Ⅱ)与其余的氧原子OS2~OS6的距离为0.267 0~0.354 8 nm。LATA等[16]研究Mn(Ⅱ)在水溶液中形成的水化膜,Mn(Ⅱ)与O的间距为0.219~0.375 nm,均存在配位作用。因而,表面氧原子对Mn(Ⅱ)存在一定的相互作用。吸附后平衡离子K+与(001)面的距离由0.155 0 nm增加到0.181 6 nm,表明K+有远离(001)面的趋势,这主要是由于在黏土矿物的基面,存在高价离子对低价离子的离子交换吸附[17]。图6B为(010)-B5构型,Mn(Ⅱ)吸附于3个≡Al—OH基团的空穴处,Mn(Ⅱ)与OS1、OS2、OS3分别形成键长为0.216 1、0.209 6、0.210 3 nm的共价键。根据LIU等[18]对2∶1型的黏土矿物端面羟基基团酸度的计算,(010)面的≡Al—OH和≡Si—OH的pHa分别为15.1、7.0。根据酸碱理论,≡Al—OH获得质子的能力强于≡Si—OH,SiO4四面体层的Si被Al取代,形成的≡Al—OH强化了对Mn(Ⅱ)的吸附。
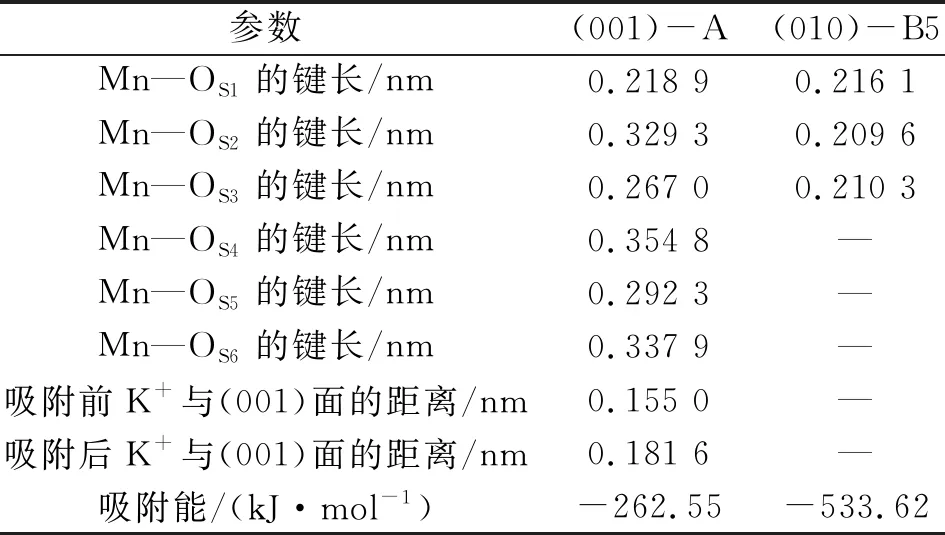
表2 稳定吸附构型参数
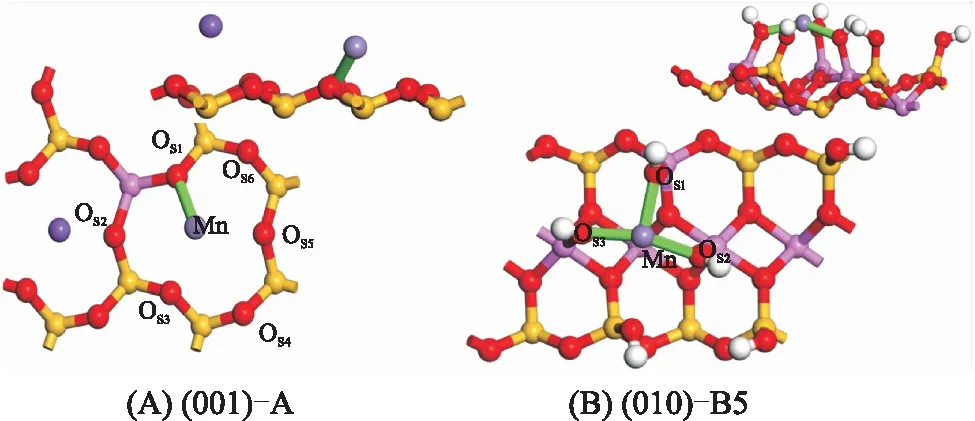
图6 Mn(Ⅱ)在伊利石表面的吸附稳定构型
Mn(Ⅱ)在(001)面和(010)面的吸附能存在较大差异,在(010)面的吸附能明显大于在(001)面的吸附能,为进一步探究吸附机理,需对稳定构型(001)-A、(010)-B5进行电荷分析和态密度分析。
2.3 电荷分析
对吸附最优构型进行电荷分析,进一步探究Mn(Ⅱ)与表面活性位间的相互作用机理。图7为吸附稳定构型的电子密度图,颜色变化表示电子密度的大小,由红色→白色→蓝色表示电子密度由小到大。图7A为(001)-A构型,电子密度图沿Mn(Ⅱ)与最近距离的2个表面OS原子的剖面,即OS1—Mn—OS3剖面,Mn(Ⅱ)的电子密度与OS1间有部分重叠,与OS3无重叠,表明Mn(Ⅱ)与OS1存在共价键作用;同样,在图7B的(010)-B5构型中,Mn—OS1—OS2—OS3的电子密度剖面显示Mn(Ⅱ)与OS1、OS2和OS3均存在电子密度的重叠,表明Mn(Ⅱ)与OS1、OS2和OS3均存在共价键作用。总之,Mn(Ⅱ)与伊利石的(001)、(010)面氧原子的电子密度均存在一定程度的重叠,从而形成共价键,这与之前的成键分析结果相吻合。
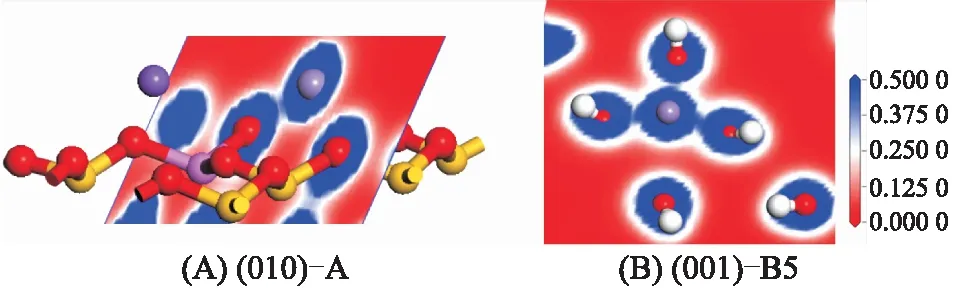
图7 稳定吸附构型的电子密度图
表3为吸附构型的Mulliken布居及电荷数,在(001)面Mn—OS1的布居为0.03,Mn—OS3和Mn—OS5的布居均为-0.01,在(010)面Mn—OS1、Mn—OS2和Mn—OS3的布居分别为0.18、0.27和0.24,布居越大表面共价键作用越强,因此在(001)面,Mn(Ⅱ)与表面氧间存在弱的共价键作用,在(010)面存在较强的共价键作用;Mn(Ⅱ)在(001)面的电荷数为2.32 e,在(010)面的电荷数为0.48 e,由于吸附体系为电中性体系,因而Mn(Ⅱ)与(001)面的静电作用明显强于(010)面。

表3 稳定吸附构型的Mulliken布居及电荷
2.4 态密度分析
态密度在相同的能量下轨道间的相互作用可表征共价键的形成[19]。图8为在表面吸附的Mn—OS的态密度,图8A为在(001)面Mn—OS1的态密度,OS1的2p轨道与Mn的4s轨道在-0.1~-0.48 eV成键,以OS1的2p轨道为主,在0~0.17 eV形成反键,以Mn的4s轨道为主。图8B为在(010)面Mn—OS2的态密度,OS2的2p轨道与Mn的4s轨道在-3.3~-11 eV成键,以2p轨道为主,在0~4.2 eV形成反键,以Mn的4s轨道为主。对比吸附在(001)面和(010)面的态密度,在(001)面Mn—OS1间的态密度峰跨度小,峰形尖锐,局域性较强,在(010)面Mn—OS2间的态密度峰跨度大,峰形平缓,离域性较强,表明在(010)面Mn与OS2间的作用强于在(001)面Mn与OS1间的作用。这与键长和布居的规律一致。Mn的4s、3d轨道均穿过费米能级,表明反应活性较强,这为解吸药剂对重金属离子的捕获提供捕捉位。
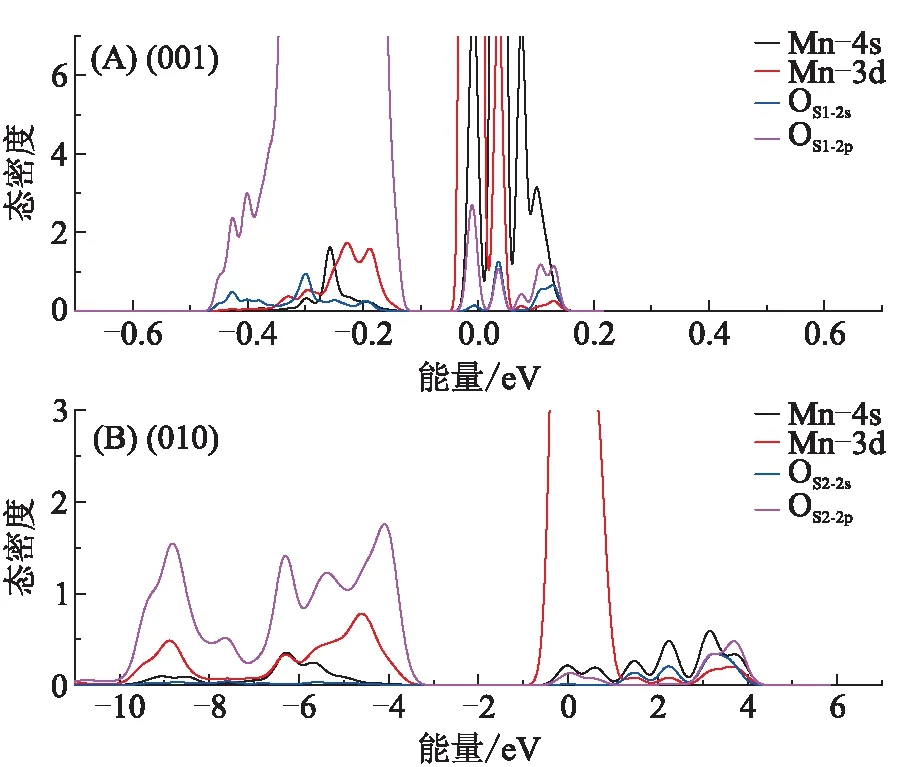
图8 表面吸附的Mn—OS态密度
3 结论
借助密度泛函理论对Mn(Ⅱ)在伊利石表面的吸附进行模拟计算与分析,从原子层面揭示吸附机理,归纳如下:
(1)在(001)面,Mn(Ⅱ)优先吸附于硅氧环空穴处,且与活性氧OS1形成1个共价键,吸附能为-262.55 kJ/mol;在(010)面,Mn(Ⅱ)与羟基基团形成1~3个共价键,随着共价键的增多,吸附能越大,吸附的最稳定构型为Mn(Ⅱ)吸附于3个≡Al—OH之间的空穴处,最优吸附能为-533.62 kJ/mol。
(2)Mn(Ⅱ)与(001)面和(010)面均存在共价键作用和静电作用,在(001)面的吸附能小于(010)面,且与(001)面以静电作用为主,与(010)面以共价键作用为主。
(3)Mn(Ⅱ)与伊利石表面共价键的形成主要是Mn的4s轨道与表面O的2p轨道间的相互作用。
(4)从吸附微观机理出发,可以通过对伊利石剥片以暴露更多的吸附位,增加对Mn(Ⅱ)的吸附;可以添加电解质以降低静电作用或通过强酸以削弱共价键作用,来完成对Mn(Ⅱ)的解吸附。
