表面硫化和磷化提升钼酸镍的析氢性能
2022-09-16姜仁政常峻华张金凤李孟江谢英鹏
姜仁政 常峻华 高 颖 张金凤 李孟江 谢英鹏*,
(1沈阳化工大学化学工程学院,沈阳 110142)(2沈阳化工大学材料科学与工程学院,沈阳 110142)
0 引 言
氢能具有能量密度大(120~142 MJ·kg-1)、热值高、转化效率高等特点,是一种理想的能源载体,也被视为21世纪最具发展潜力的清洁能源[1-3]。目前H2的主要来源是通过化石燃料制取的“灰氢”,生产过程伴随着大量二氧化碳(CO2)排放。利用可再生能源产生的电能驱动水分解反应产生H2,不仅可以获得“绿氢”(过程无CO2排放),还可以解决可再生能源的存储和运输问题,是最理想的能源利用方式[4-5]。电解水析氢反应(HER)需要引入合适的催化剂来降低反应过电势,提高析氢效率。贵金属Pt是最高效的HER催化剂,然而Pt的稀缺和价格昂贵限制了电解水制氢技术的大规模应用[6-7]。因此,开发储量丰富、经济高效的电解水催化剂是生产“绿氢”的关键,同时也面临着巨大挑战。
过渡金属Mo/Ni基硫化物或磷化物因具有优异的电催化性能,成为有望替代贵金属的HER催化剂。研究发现,通过构建异质结构,并结合掺杂、形貌调控和缺陷工程等方法,可以显著提高Mo/Ni基化合物的HER性能。例如,Mo掺杂Ni3S2/NixPy形成丰富的掺杂异质结构,通过协同优化OH*、O*和OOH*中间物种的生成吉布斯自由能,加快了电化学分解水反应动力学速率[8]。泡沫镍(NF)负载MoS2/Ni3S2具有高活性异质界面,利于电荷传递,提升HER和析氧反应(OER)动力学和反应活性[9]。高分散Mo原子掺杂具有丰富氧缺陷结构的Ni2P/NiMoO4-y,提供了更多的HER协同活性位点,提高了导电性,促进了传质过程[10]。3D分级异质结构MoP/Ni2P自支撑电极,因其结构特点以及磷化物组分间的协同作用,拥有优越的分解水性能[11]。此外,相比于晶相材料,非晶相电催化剂材料(如非晶相纳米片结构CoMoS[12])具有短程有序、长程无序的特点,其较大的比表面积可以为HER过程提供大量的开放活性位点。
Mo/Ni基氧化物,如钼酸镍(NiMoO4),也是被广泛研究的HER催化剂。虽然NiMoO4自身HER性能有限,但通过如前所述的各种改性方法也可大幅提高其电催化活性。例如,在前期研究中我们发现,P掺杂和磷酸盐负载的NiMoO4具有优异的HER性能[13],这主要是因为P掺杂提高了NiMoO4的导电性,而表面负载的磷酸盐提供了丰富的表面反应位点。
我们首先制备了NiMoO4纳米阵列自支撑泡沫镍电极(NMO/NF),再通过水热硫化法在其表面形成非晶相硫化物壳层,最后通过气相磷化法进行表面处理获得硫化物和磷酸盐共同修饰的PS-NMO/NF电极。由于具有非晶相硫化物和磷酸盐,PS-NMO/NF电极的表面活性位大幅增加,而且硫化物与磷酸盐之间形成的异质界面利于电子传输,从而大幅提高电极的分解水析氢性能。结果表明,在1 mol·L-1KOH电解液中,电流密度为10和100 mA·cm-2时,PS-NMO/NF所对应的析氢过电势分别为93和180 mV,并展现出优异的长时稳定性。
1 实验部分
1.1 原料与试剂
NF导电基体购自昆山佳胜电子有限公司,厚度为1.5 mm,每平方厘米孔数为17个;四水合钼酸铵((NH4)6Mo7O24·4H2O)、六水合硝酸镍(Ni(NO3)2·6H2O)、水合次亚磷酸钠(NaH2PO2·H2O)、氢氧化钾(KOH)、硫代乙酰胺(CH3CSNH2)均为分析纯,购自国药控股化学试剂有限公司;盐酸(HCl)、丙酮(CH3COCH3)、无水乙醇(CH3CH2OH)均为分析纯,购自天津大茂化学试剂有限公司;去离子水(18.2 MΩ·cm)采用德国Millipore系统制备。
1.2 电催化剂的制备
PS-NMO/NF制备过程如图1所示,首先通过水热过程制备NMO/NF,再通过二次水热硫化过程制备硫化改性的NMO/NF(S-NMO/NF),最后通过磷化过程获得PS-NMO/NF。具体步骤如下:
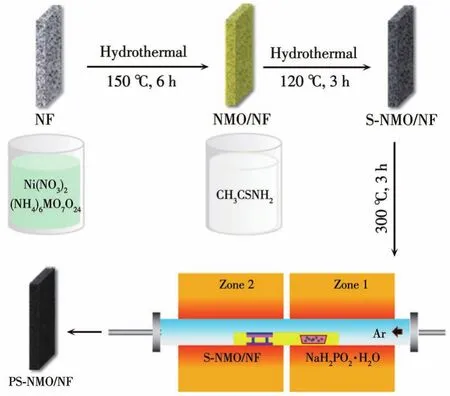
图1 PS-NMO/NF的制备流程示意图Fig.1 Schematic diagram of the preparation process of PS-NMO/NF
制备NMO/NF:按照前期工作中的合成方法[13],首先配制15 mL(NH4)6Mo7O24(15 mmol·L-1)和Ni(NO3)2·6H2O(35 mmol·L-1)的水溶液;随后将上述均一溶液转入20 mL的聚四氟乙烯反应釜内胆中,并将预处理的NF(3 mol·L-1HCl超声清洗30 min,然后分别用丙酮、乙醇和去离子水超声清洗3次)斜靠于内胆壁;将内胆装入反应釜密封后,在烘箱中150℃下反应6 h。反应完成后取出NF电极,经去离子水和无水乙醇多次清洗,60℃干燥过夜后得到NMO/NF电极。此外,反应釜内胆中生成的粉末样品同样经过清洗和干燥,获得NMO样品。
制备S-NMO/NF:将NMO/NF电极置于15 mL硫代乙酰胺(2.6 mmol·L-1)的水溶液中进行二次水热处理。在烘箱中120℃下反应3 h,经去离子水和无水乙醇多次清洗,60℃干燥过夜后得到S-NMO/NF电极。将NMO粉末经过同样水热处理过程,经清洗干燥后获得S-NMO粉末样品。
制备PS-NMO/NF:将S-NMO/NF置于高温管式炉下游,上游放置2.0 g NaH2PO2·H2O粉末,在Ar气氛中(流量为40 mL·min-1)300℃(升温速率为3℃·min-1)热处理3 h,得到PS-NMO/NF电极,PS-NMO负载量约为7.2 mg·cm-2。将S-NMO粉末经过同样磷化处理,获得PS-NMO粉末。
1.3 电催化剂的表征方法
用日本理学的D/max-2500 PC型X射线衍射仪(XRD)分析物相组成,辐射源为 CuKα1射线,λ=0.154 18 nm,管电压为50 kV,管电流为200 mA,扫描 步幅为 0.02°,2θ=10°~80°。用德 国 Zeiss的SUPRA 55 SAPPHIRE场发射扫描电子显微镜(SEM)表征电极的微观形貌,用配置的牛津OXFORD X射线电制冷能谱仪(EDS)分析元素组成和分布,加速电压为5~30 kV。用高分辨透射电镜(HRTEM,TECNAI G20)表征电催化剂的微观结构和晶格结构,测试用粉体样品是通过超声剥离所制备电极获得。用美国Thermo的ESCALAB 250型X射线光电子能谱(XPS)分析样品表面的元素组成和价态,辐射源为AlKα射线(1 486.6 eV),所有键能采用污染碳C1s轨道键能(284.6 eV)进行校准。
1.4 电化学实验
采用单槽三电极体系,以自制的自支撑电极为工作电极、石墨棒为对电极、Ag/AgCl电极为参比电极,在1.0 mol·L-1KOH电解液中进行电化学实验。利用CHI 660E电化学工作站进行数据采集。所有电势采用可逆氢电极电势(RHE)进行校准(ERHE=EAg/AgCl+0.204 6+0.059 1pH),电流密度基于电极的反应几何面积(1 cm2)进行计算。在自动IR补偿(90%)模式下,扫描速率控制在2 mV·s-1,采用线性扫描伏安法(LSV)测得HER极化曲线。然后,根据Tafel方程(η=blg|j|+a,其中η是过电位,b是Tafel斜率,j是电流密度)和线性拟合获得Tafel斜率。采用不同扫描速率下的循环伏安(CV)曲线测得双电层电容(Cdl)和电化学活性表面积(ECSA)。利用电化学阻抗谱(EIS)技术在析氢过电位100 mV下测定电荷传递阻力,扫描范围为100 kHz~0.01 Hz,振幅为5 mV。运用恒电流技术考察电催化剂的稳定性。
2 结果与讨论
2.1 电催化剂的表征
采用XRD分析电催化剂的物相组成和晶体结构。由于自支撑电极材料中基体Ni骨架含量较多,造成钼酸镍及其表面生长的活性组分特征峰较弱,难以分析。因此,采用NMO、S-NMO和PS-NMO粉末进行XRD分析,结果见图2。由图可知,位于2θ=14°、26°、27°和34°处的衍射峰可归属于β-NiMoO4的特征峰(PDF No.12-0348),在29°出现的微弱衍射峰属于α-NiMoO4(PDF No.33-0948)。而在 2θ=10°(插图)、21°、22°、27°、30°和 31°的衍射峰对应 NiMoO4·H2O(PDF No.13-0128)[14-15]。通过比较3个样品的XRD图可以看出,NMO主要由β-NiMoO4和NiMoO4·H2O两相构成,同时含有极少量的α-NiMoO4。依次经液相硫化和气相磷化处理后,得到的S-NMO和PS-NMO粉体仍然以混合相NiMoO4为主,未发现硫化物和含磷化合物的特征峰存在,这表明硫化、磷化作用仅能在NiMoO4表层生成含量很少的硫化物和磷化物。
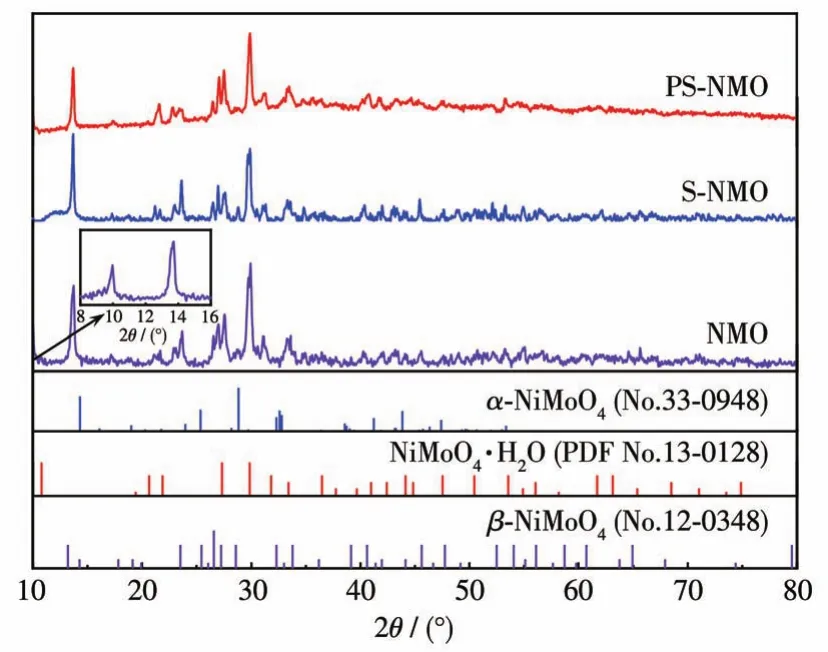
图2 NMO、S-NMO和PS-NMO的XRD图Fig.2 XRD patterns of NMO,S-NMO,and PS-NMO
图3a为NMO/NF的SEM照片,由图可见,在NMO/NF中约5 μm长的NMO纳米棒以团簇形式生长在NF基体上。不同倍数下PS-NMO/NF的SEM照片(图3b、3c)显示,NMO/NF依次经硫化、磷化处理后,棱角分明的纳米棒团簇结构转变成表面粗糙的类珊瑚球状结构,表明在NMO表面有非晶相生成。我们在前期工作中发现,磷化作用会在NMO表面生成大量非晶相含磷物种,但不会明显改变NMO的纳米棒形貌[13]。因而,PS-NMO/NF的结构变化应该主要归功于硫化作用。非晶相类珊瑚球状多级结构保持了良好的传质扩散性能,而且提供了大量的活性位点,将有助于提高PS-NMO/NF的催化活性。EDS元素分布图(图3d~3h)显示,PS-NMO/NF表面含有Ni、Mo、O、S和P元素,而且呈现均匀分布。

图3 NMO/NF(a)和PS-NMO/NF(b、c)的SEM照片;(c)中矩形部分的元素分析图(d~h)Fig.3 SEM images of NMO/NF(a)and PS-NMO/NF(b,c);Corresponding element mapping images(d-h)in the rectangular area of(c)
图4为自支撑电极表面S-NMO和PS-NMO粉体的TEM和HRTEM照片。由图4a可见,棒状结构的S-NMO表层覆盖有一层不均匀厚度的薄层。如图4b所示,该薄层主要由无定形相构成,并在无定形相中分散着结晶度不高的结晶相。根据晶面间距0.24 nm判断该物种应属于多硫金属化合物,但在XRD图中无明显衍射峰出现,因此该物相尚不清楚。另外,在S-NMO的HRTEM照片中,可以看到在体相中有晶面间距为0.88 nm的晶格条纹存在,根据标准卡片PDF No.13-0128可知,该晶格结构对应NiMoO4·H2O在10°处的衍射峰,与文献报道相一致[15]。S-NMO经300℃磷化处理后,得到的PS-NMO仍然保持着棒状结构(图4c)。然而P的掺入导致了表层钼酸镍晶格形变,以致结构扭曲的晶格条纹无序地散落在无定形相的薄层中(图4d),该晶面间距为0.65 nm,根据PDF No.12-0348推断,这归属于β-NiMoO4位于14°处的衍射峰。另外,在体相中可以清晰地观察到晶面间距为0.47 nm的晶格条纹,该条纹属于β-NiMoO4位于19°处的衍射峰。由以上结果可知,PS-NMO电催化剂体相主要由NiMoO4·H2O和β-NiMoO4构成,经硫化、磷化处理后表面形成以非晶相为主的外延活化层。外延活化层中无定形物相与散落在其中的且结晶度不高的晶相构成了丰富的异质界面。该异质结构与变形的晶格结构为电解水析氢过程提供了丰富的活性位点[16-18]。

图4 S-NMO(a、b)和PS-NMO(c、d)的TEM和HRTEM照片Fig.4 TEM and HRTEM images of S-NMO(a,b)and PS-NMO(c,d)
采用XPS进一步分析了NMO、S-NMO、PS-NMO粉体的表面组成和化学价态,结果见图5。如XPS全谱图所示(图5a),与NMO相比,S-NMO和PS-NMO表面除了含有Mo、Ni、O元素之外,还分别增加了S和S、P元素,进一步证明S、P元素与NMO表面发生了相互作用,并存在于NMO表面,该结果与EDS相一致。PS-NMO的P2p谱图(图5b)显示,位于133.9和134.8 eV处的特征峰分别归属于磷酸盐中P—O键的P2p3/2和P2p1/2轨道[19],而在129 eV左右无峰出现,表明P掺杂到S-NMO中,主要形成了磷酸盐。S-NMO的S2p谱图(图5c)显示,在168.7处的宽峰属于S—O键[20],表明S进入了NMO晶格形成S—O—M键;位于161.7、162.9 eV和163.0、164.2 eV处的2组主峰(峰间距为1.2 eV),分别对应于M—S键和M—Sx键的S2p3/2和S2p1/2轨道[21-22],表明在硫化过程中S元素与NMO发生了表面反应,生成金属硫化物和多硫化物。在进一步磷化后,多硫化物主峰消失,且M—S键的特征峰向低键能方向移动0.2 eV,表明在磷化过程中NaH2PO2·H2O分解产生的H3P与硫化物和多硫化物发生相互作用,将多硫化物中富余的S移除,并使高价金属还原,导致M—S键能红移。该结果证明了P与硫化物间存在相互作用,并形成了异质结构。O1s谱图(图5d)展现出相似的变化规律。如NMO的O1s谱图中位于530.8 eV处的特征峰为M—O键的特征峰[22-23],在532 eV处的特征峰对应H—O键[19,23]。在硫化过程中,硫化作用将NMO表面羟基脱除生成硫化物,并形成吸附氧物种,对应S-NMO的O1s谱图中533.0 eV处特征峰[19]。而且,在531.4 eV处出现了明显的缺陷氧(Ov)特征峰[23],表明在NMO表面形成了大量具有金属配位不饱和的硫化物,其具有非晶相的特征。在高温水热条件下,硫代乙酰胺发生水解生成H2S。H2S将表面金属还原引起M—O键特征峰向低能态方向迁移0.2 eV。在进一步磷化作用下,该缺陷氧结构倾向于吸附PO43-基团,消耗了表面氧缺陷,生成磷酸盐[13,24],因此在532.5 eV处出现PO43-基团的P—O键特征峰[25]。另外,P掺入NMO晶格使M—O键特征峰发生蓝移,而S—O键及表面吸附氧物种因发生磷化还原和脱硫反应被移除,该现象与S2p谱图结果相对应。
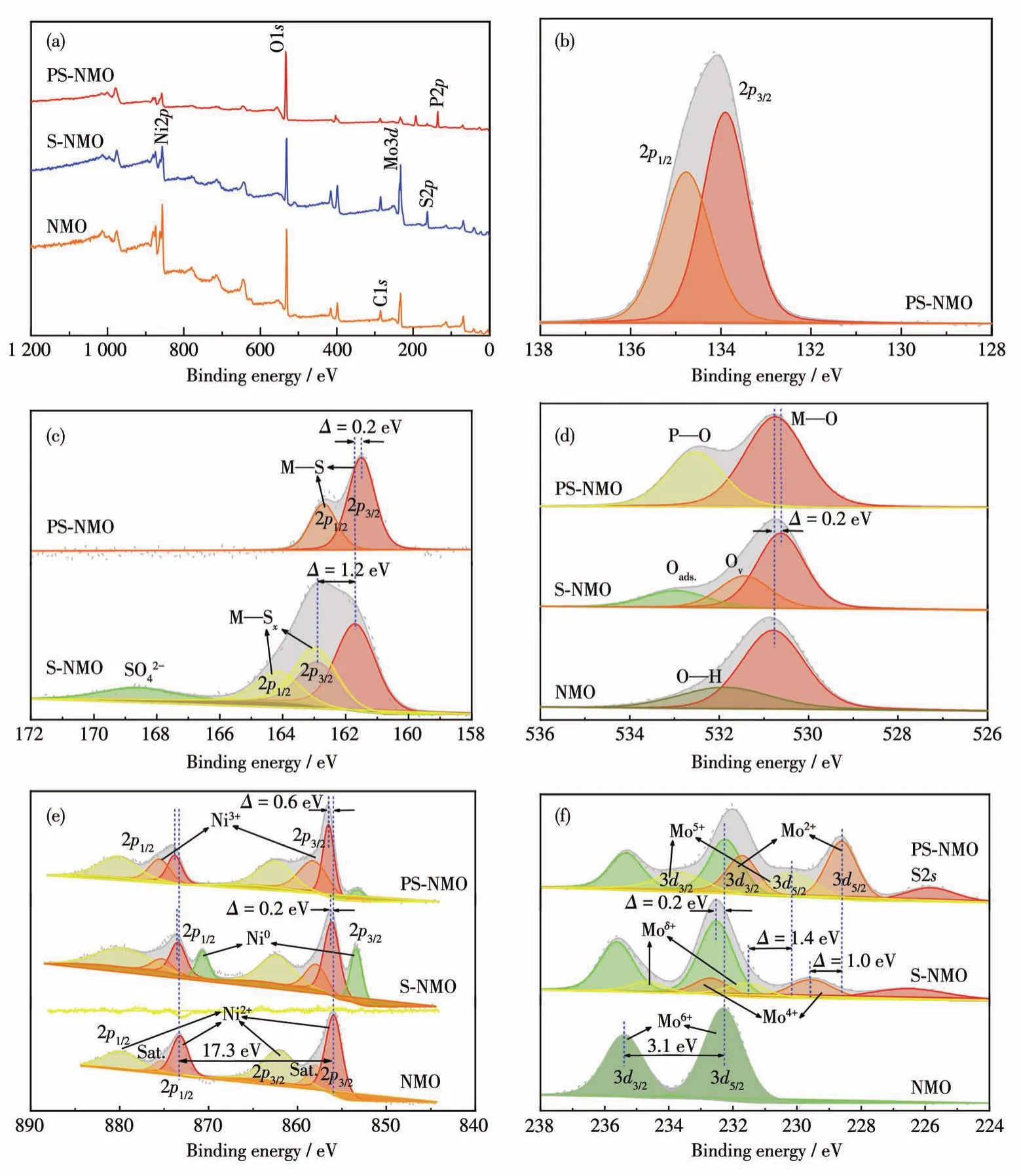
图5 NMO、S-NMO和PS-NMO粉末的XPS全谱图(a)、P2p(b)、S2p(c)、O1s(d)、Ni2p(e)和Mo3d(f)谱图Fig.5 XPS survey(a),P2p(b),S2p(c),O1s(d),Ni2p(e)and Mo3d(f)spectra of NMO,S-NMO,and PS-NMO powders
如上所述,在硫化和磷化过程中主要发生成盐反应和还原反应,这2个过程在Ni2p和Mo3d谱图中也得以体现。如图5e所示,NMO在855.9、862.1 eV和873.2、879.4 eV处出现的2组峰分别属于Ni2p3/2和Ni2p1/2的主峰及卫星峰,且2个主峰间的能级间距为17.3 eV,证明NMO晶格中Ni主要以Ni2+为主[11,26],而在858和875.3 eV处出现的特征峰证明有Ni3+存在[19,27]。在硫化过程中,由于S进入NMO晶格,导致Ni2+和Ni3+特征峰整体向高键能方向迁移0.2 eV。同时,在853.4和870.7 eV处出现明显的Ni0特征峰[11],证明表面硫化作用产生大量的硫化镍化合物。但在进一步磷化作用下,Ni0含量显著下降,而Ni3+含量显著增加,且Ni2+和Ni3+特征峰进一步向高键能方向移动0.4 eV,结合S2p和O1s谱图,进一步表明PS-NMO中有高价态磷酸镍物种生成,进而证明硫化物/磷酸盐异质结构的形成。图5f为Mo3d谱图,由图可见,在NMO中位于232.3和235.4 eV处的峰间距为3.1 eV的2个主峰分别归属于Mo6+的3d5/2和3d3/2[11,13,28-30],证明在NMO中只有Mo6+存在。经硫化处理后,在NMO表面部分Mo6+被硫代乙酰胺水解产物H2S还原,导致在231.6、234.7 eV和229.5、232.6 eV处分别出现Moδ+(5<δ<6)和Mo4+。而且,S掺入NMO晶格中,形成S—O—Mo键使Mo6+的特征峰发生蓝移,此外在226.6 eV处S2s特征峰的出现也证明有硫化物生成[8]。在磷化作用下,H3P还原诱导Mo向低价态转变,并向低键能方向迁移0.2 eV,而形成的Mo5+和Mo2+的含量明显增加[13,29-30]。Mo元素的价态变化同样证明了磷化作用对硫化物的影响,并进一步证实硫化物/磷酸盐异质结构的形成。综上所述,硫化、磷化作用得到的PS-NMO表面具有丰富的硫化物和磷酸盐物种,构成了丰富的异质结构界面。
2.2 电化学性能测试
图6a为NF、NMO/NF、S-NMO/NF和PS-NMO/NF在室温下1 mol·L-1KOH溶液中的HER性能。从图中可见,NMO/NF由于具有较差的导电性(尽管样品表面经过喷金处理,但是NMO/NF的SEM图像仍存在明显的放电现象),表现出最差的HER活性[13,31-32]。通过液相硫化改性后形成的二元金属硫化物具有低电导率,且Ni和Mo元素间协同作用使NiMoSx物种在边缘Mo位点的H原子吸附自由能接近于0[33],此外硫化后得到的具有粗糙表面的类珊瑚球结构,提供了大量开放的活性位点,从而显著提高了HER活性。当电流密度为10 mA·cm-2时,HER过电势从NMO/NF的240 mV降至约100 mV(S-NMO/NF)。进一步经磷化处理后,P元素以磷酸盐和掺杂形式存在于PS-NMO/NF中。磷酸盐物种可作为质子载体,加速质子传递[33],而且磷原子掺入硫化物中形成的M—P—S键,能够提高活性位点数量,降低电子结合能,促进电子转移,进而提高HER活性[34]。因此,PS-NMO/NF的HER活性进一步增加,当电流密度为10 mA·cm-2时,HER过电势约为93 mV,而且在100 mA·cm-2时,仅为180 mV,该HER性能显著好于相对应的S-NMO/NF和P-NMO/NF电催化剂(电流密度为10 mA·cm-2时,HER过电势为148 mV[13]),并与商用催化剂性能相近[35]。图 6b为 NF、NMO/NF、S-NMO/NF和PS-NMO/NF的Tafel曲线图,可以发现PS-NMO/NF的Tafel斜率为67 mV·dec-1,明显低于NMO/NF(116 mV·dec-1)、NF(101 mV·dec-1)、S-NMO/NF(79 mV·dec-1)和 P-NMO/NF(88 mV·dec-1[13])。根据HER的Tafel理论值可知,这些电极的HER过程都遵循Volmer-Heyrovsky机制[36-37],而PS-NMO/NF显示出较快的HER速率。
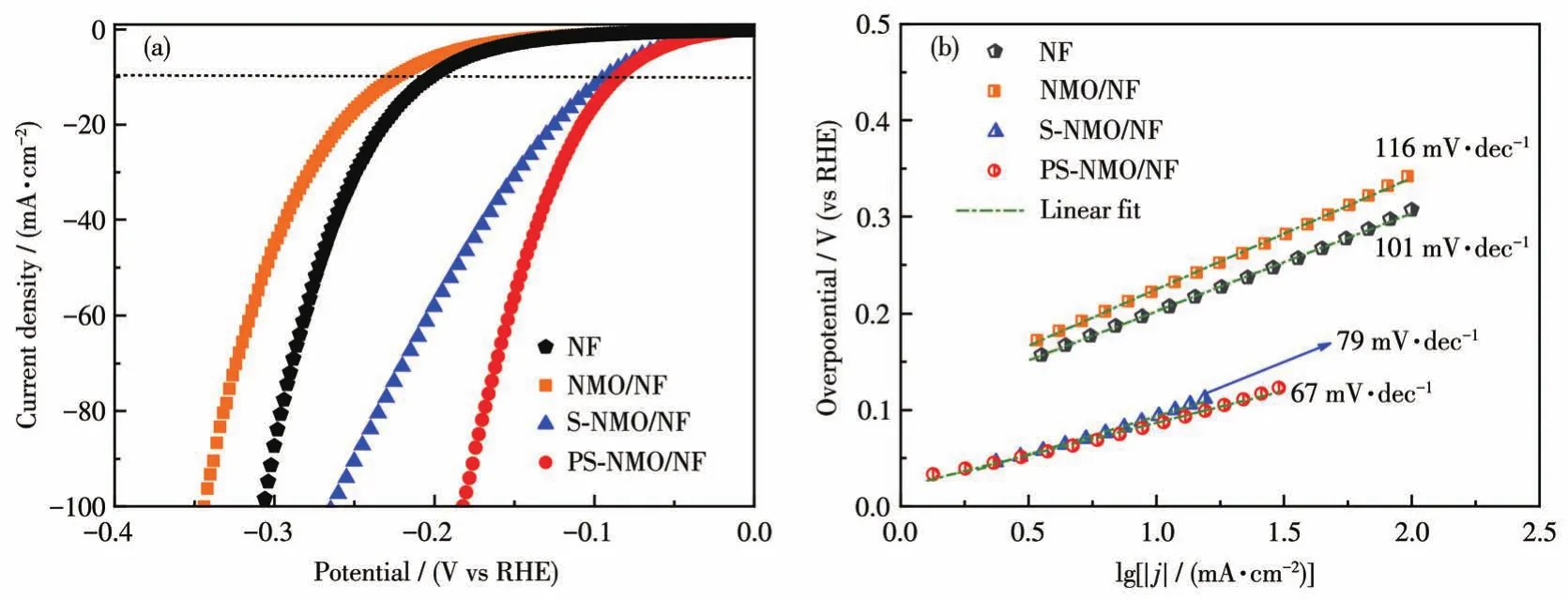
图6 NF、NMO/NF、S-NMO/NF和PS-NMO/NF在1 mol·L-1KOH中的LSV曲线(a)和Tafel曲线(b)Fig.6 LSV curves(a)and Tafel slopes(b)of NF,NMO/NF,S-NMO/NF,and PS-NMO/NF in 1 mol·L-1KOH
采用CV法测定电极的Cdl值,Cdl值可反映电极的ECSA大小[38-39]。图7a、7b分别为S-NMO/NF和PS-NMO/NF在非法拉第区域、不同扫描速率下的CV图,再由CV数据计算出各样品的Cdl值。由图7c可知,S-NMO/NF催化剂的Cdl值高达333.88 mF·cm-2,远远高于NMO/NF电极的0.67 mF·cm-2。在前期工作中,尽管有大量的非晶相存在,P-NMO/NF的Cdl值仅从NMO/NF电极的0.46 mF·cm-2增加到36.2 mF·cm-2[13]。因此,S-NMO/NF催化剂具有如此高的Cdl值可证明在其表面有大量的非晶相硫化物形成,该结果与SEM和XPS结果相一致。与S-NMO/NF相比,PS-NMO/NF催化剂的Cdl值下降,但仍然高达255.08 mF·cm-2,这是由于在300℃磷化作用下,催化剂表面不稳定含硫物种和易挥发物质被移除以及部分非晶相颗粒发生凝聚。尽管PS-NMO/NF催化剂的Cdl值有所下降,但其HER活性明显增加,表明磷化作用提高了S-NMO/NF的本征HER性能。
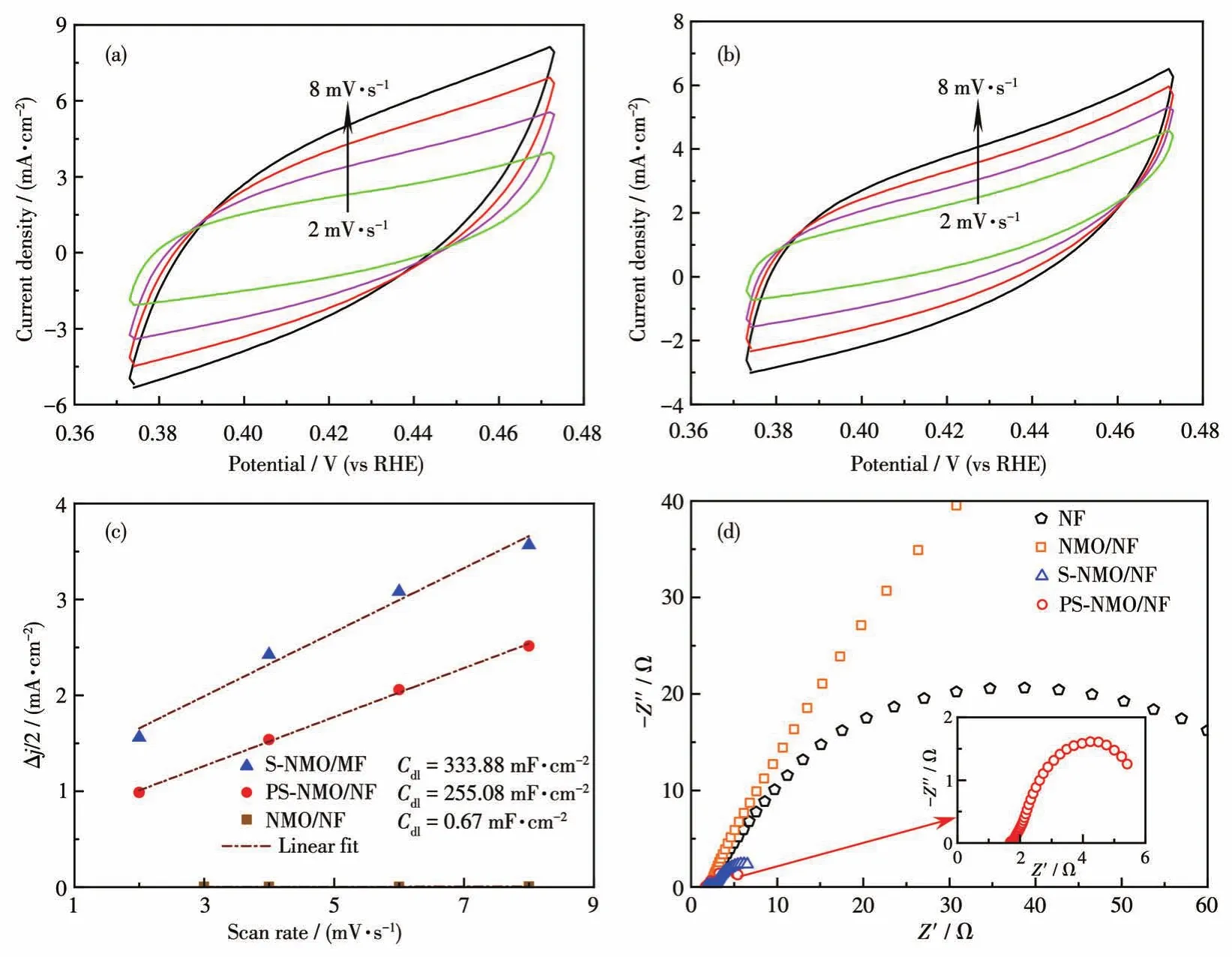
图7 S-NMO/NF(a)、PS-NMO/NF(b)的CV曲线;NMO/NF、S-NMO/NF和PS-NMO/NF的电流密度随扫描速率的变化曲线(c);NF、NMO/NF、S-NMO/NF和PS-NMO/NF的EIS谱图(d)Fig.7 CV curves of S-NMO/NF(a)and PS-NMO/NF(b);Plots of current density as a function of scan rates for NMO/NF,S-NMO/NF,and PS-NMO/NF(c);EIS spectra of NF,NMO/NF,S-NMO/NF,and PS-NMO/NF(d)
在过电势100 mV下对NF、NMO/NF、S-NMO/NF和PS-NMO/NF进行EIS测试。如图7d所示,根据Nyquist曲线和EIS拟合数据可知,这些样品具有相近的溶液电阻(Rs),约为2.0 Ω,而电荷传递电阻(Rct)差距明显。其中,NMO/NF 的Rct值最高(280 Ω),表明了NMO具有较差的导电性,抑制了HER活性[13,31-32]。采用液相硫化处理后,S-NMO/NF的Rct值急剧下降(约7 Ω),证实NMO/NF表面生成的多元硫化物大幅度提高了电荷传输效率。再经磷化改性后,获得的PS-NMO/NF催化剂的Rct值进一步下降至5 Ω,证实了P原子的引入提高了S-NMO/NF导电性,加快了电荷传递速率。采用恒电流技术考察了PS-NMO/NF在1 mol·L-1KOH中的HER稳定性。如图8a所示,在20 h内电流密度没有明显衰减,而且稳定性测试后的HER性能显示当电流密度分别为10和100 mA·cm-2时,过电势仅增加10和20 mV(图8b),并且从插图中可知,反应后的Rct值也没有明显增加,表明该催化剂具有良好的稳定性。
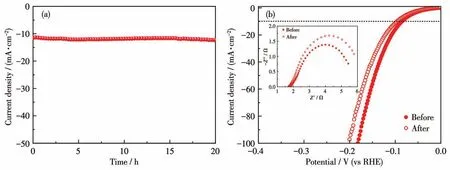
图8 PS-NMO/NF在1 mol·L-1KOH中的I-t曲线(a)以及HER循环后的LSV曲线和EIS谱图(插图)(b)Fig.8 (a)I-t curve,(b)LSV curves,and EIS spectra(Inset)after HER cyclic test of PS-NMO/NF in 1 mol·L-1KOH
3 结 论
先后采用水热硫化和气相磷化法处理钼酸镍纳米棒阵列,获得具有类珊瑚球结构的自支撑PS-NMO/NF电极。硫化作用使钼酸镍纳米棒表面生成非晶相硫化物,显著提高了电化学活性面积,增加了活性位点,并提高了导电性。磷化处理过程使P元素引入硫化物中并形成非晶相磷酸盐,构建了丰富的硫化物/磷酸盐异质界面,降低了电荷传递阻力,促进了电子转移,提升了HER性能。在1 mol·L-1KOH电解液中,电流密度分别为10和100 mA·cm-2时,PS-NMO/NF电极的HER过电势分别为93和180 mV,Tafel斜率为67 mV·dec-1,而且在20 h内可稳定运行。该工作为开发廉价、高效且具有丰富异质界面的电催化剂提供了新思路。
