基于小分子化合物库筛选协同多腺苷二磷酸核糖聚合酶抑制剂杀伤卵巢癌细胞的药物
2022-09-07李含笑李小满罗业飞王嘉东
李含笑, 李小满, 罗业飞, 王嘉东
(北京大学基础医学院放射医学系,系统生物医学研究所,北京 100191)
全球每年约有24万例新的卵巢癌病例被确诊,尽管采取了手术和铂类药物化疗这两种目前常用的治疗手段,但是仍然有超过15万名女性死于该疾病,因此迫切需要找到新的治疗手段[1]。近年,随着单细胞测序技术的迅速发展,卵巢癌的肿瘤异质性已经逐渐被人们熟知,这就为靶向治疗的应用提供了依据。目前,已知的参与DNA双链断裂损伤修复的同源重组修复(homologous recombination repair,HRR)蛋白乳腺癌易感基因1(breast cancer susceptibility gene 1,BRCA1)和 乳腺癌易感基因2(breast cancer susceptibility gene 2,BRCA2),在卵巢癌中占有较高的比例,大约有50%的高级别浆液性卵巢癌患者带有HRR的缺陷[2]。根据Gao等人对中国卵巢癌胚系BRCA基因变异图谱的分析,中国卵巢癌的整体人群突变率为21.79%,这一数字远高于国外卵巢癌的BRCA基因突变发生率(13%)[3]。它们的突变可以导致细胞基因组不稳定性的增加,从而导致一些抑癌基因的激活或者染色体的融合、杂合缺失等现象,最终导致BRCA基因突变携带者患癌风险显著增加[4]。
HRR 功能缺陷的细胞对铂类药物和新开发的 PARP 抑制剂都具有敏感性[5]。HRR 功能缺陷的细胞对PARP抑制剂敏感性增加的现象是经典的“合成致死”效应。2014年,奥拉帕尼(olaparib)成为第一个被批准用于治疗癌症的PARP抑制剂[6]。当PARP 抑制剂单用时,可通过合成致死选择性地靶向具有BRCA1 或BRCA2基因突变的肿瘤细胞。然而,当与其他治疗药物一起使用时,PARP 抑制剂也显示出相当大的应用前景。3种PARP抑制剂被批准用于治疗卵巢癌:Olaparib[7]、卢卡帕尼(rucaparib)[8]和Niraparib[9]。PARP抑制剂现在也进入一线卵巢癌的治疗研究,尤其是最新开发出来的PARP抑制剂Niraparib,其不仅对HRR缺陷的卵巢癌病人有较好的治疗作用,而且对HRR阳性的卵巢癌患者具有良好的治疗作用[10]。其他HR修复能力正常的卵巢癌患者通常对单一的PARP抑制剂表现出不敏感。此外,有些细胞会对PARP抑制剂产生耐药,例如离子泵相关蛋白质的高表达导致药物外流增加[11]、PARP1 捕获能力的下降[12]、HRR功能的恢复[13]和复制叉稳定性的增加[14,15]等。因此,这些患者代表了一个相当大的群体,其临床需求尚未得到满足。
将 PARP抑制剂与能够降低 HR修复的药物相结合可能会使这些 HR修复能力正常的卵巢癌人群变得敏感[16]。HR修复通路是一种多成分途径,据报道,一些靶向该途径成分的药物可能会抑制 HR修复。目前,已经有许多药物联合使用治疗肿瘤的成功案例[17-19],其中一些可以诱导 HR缺陷的表型,从而使癌症患者对 PARP抑制剂敏感。因此,有必要寻找新的与PARP抑制剂具备协同杀伤肿瘤细胞作用的新药物,从而为临床治疗卵巢癌等肿瘤提供临床前的参考依据。本文通过对379种抗肿瘤小分子化合物进行初步筛选,最终获得8个潜在的与PARP抑制剂具有协同杀伤作用的小分子化合物,本文对其中的TrKA激酶抑制剂与PARP抑制剂协同杀死卵巢癌细胞的分子机制进行了初步探究。
1 材料与方法
1.1 试剂和抗体
379种抗肿瘤活性小分子化合物购自Selleck公司,PARP抑制剂Niraparib由再鼎医药(上海)有限公司提供,其他常规化学试剂均购自北京通广有限公司。γH2AX抗体(2577 S)购自Cell Signaling Technology公司,RAD51抗体由浙江大学黄俊教授提供,53BP1抗体(612522)购自BD公司。
1.2 细胞培养
人卵巢癌细胞系SKOV3、OVCAR-8,人结肠癌细胞系HCT116, 人宫颈癌细胞系HeLa,人骨肉瘤细胞系U2OS均为本实验室传代培养,使用含有10%胎牛血清和1%青霉素/链霉素双抗生素的DMEM培养基培养,培养条件为37 ℃,5% CO2浓度。
1.3 CCK8检测
每组细胞6个复孔,3个复孔作为0 h时间点的A测量值,3个复孔作为96 h时间点A测量值;加入10 μL CCK8;培养2 h,测定A450吸光度值,作为0 h时间点值;剩下的细胞继续培养96 h,再加入10 μL CCK8,孵育2 h,测定A450吸光度值,作为96 h时间点值。之后分析合成致死的结果,利用CalcuSyn软件中的Chou-Talalay方程计算2种化合物的药物联合指数(combination index,CI),该方程考虑了半数抑制浓度(median inhibition concentration,IC50)和剂量-效应曲线的形状[20]。CI<1表示协同作用,CI=1表示加和作用,CI>1表示拮抗作用。
1.4 考马斯亮蓝染色检测细胞增殖
按照Zamoner 等人的方法进行细胞增殖检测[21]。SKOV3卵巢癌细胞以1×105个/mL细胞接种于35 mm培养皿,在药物处理前2 h更换培养基。处理分为4组:DMSO处理组,2.25 μmol/L的Niraparib处理组,6.3 μmol/L的TrKA抑制剂GW441756 处理组以及2种药物的联合处理组。之后每隔24 h用考马斯亮蓝染色观察细胞的生长情况,并拍照留存。
1.5 免疫荧光分析
为了检测DNA损伤修复蛋白质在细胞内的定位变化,本文在不同处理条件下使用免疫荧光检测细胞的损伤或者修复情况。具体如下:在35 mm铺有盖玻片的细胞皿中铺板,当细胞融合度达到30%~40%时进行相应的处理;吸掉培养基,PBS洗涤1次,加入预冷的4%多聚甲醛,固定15 min;PBS洗涤细胞3次,加入1 mL终浓度为0.25%的Triton 100处理5 min;PBS洗涤3次,加入1 mL 终浓度为2%的BSA室温封闭5 min;加入一抗(2% BSA稀释),室温孵育1 h;PBS洗涤细胞3次,加入二抗(2% BSA稀释),室温避光孵育30 min;PBS洗涤细胞3次,加入DAPI(1∶10 000,PBS稀释)染核5 min;PBS洗涤细胞3次,封片剂封片,共聚焦显微镜下分析结果。统计每个细胞中 γH2AX 和 RAD51 foci的数量,并使用 GraphPad Prism绘制数据。
1.6 克隆形成实验
将细胞梯度稀释至密度为1 200个/mL;铺板,每个6 cm培养皿加入1 mL梯度稀释好的细胞,再补加3 mL新鲜培养基,振荡使细胞分布均匀,培养8~12 h;分为DMSO处理组,GW441756处理组(处理48 h后更换新鲜培养基),Niraparib处理组(处理24 h后更换新鲜培养基),以及联合处理组(先用含有GW441756的培养基培养12 h,再加入Niraparib 培养24 h,最后用仅含有GW441756的培养基培养48 h)。总共培养10~14 d后弃掉培养基,考马斯亮蓝染色30 min,清水脱色,晾干,拍照及统计克隆数,相对存活率=处理组实验细胞克隆数/非处理组实验细胞克隆数×100%。
1.7 彗星电泳检测
取180 μL(质量体积比为0.5%)的琼脂糖液均匀滴加到载玻片上,加盖玻片,4 ℃放置10 min待琼脂糖胶凝固后取下盖玻片;将一定量的单细胞悬液与1%的低熔点琼脂糖等体积混合后加到琼脂糖垫上;加上盖玻片,4 ℃放置10 min,待琼脂糖胶凝固;将制备好的凝胶载玻片平放于平皿中,去掉盖玻片,加入预冷的细胞裂解液(2.5 mol/L NaCl,0.1 mol/L EDTA-2Na,10 mmol/L Tris-HCl,pH=10.0,10% DMSO,1% Triton X-100),4 ℃裂解过夜;用双蒸水洗涤1次;加入新配制的电泳缓冲液(300 mmol/L NaOH,1 mmol/L EDTA-2Na,pH=13.0),平衡0.5~1 h;之后用20 V,200 mA,低温条件下电泳30 min;将载玻片用中和液(0.4 mol/L Tris-HCL,pH=7.50)中和2次,每次15 min; 滴加100 μL PI避光染色10 min;PBS洗涤载玻片1次,荧光显微镜下拍照,用Image J软件统计尾部运动的长度。
1.8 放射线照射实验
提前8~12 h进行细胞铺板,将细胞放入X射线辐射仪中,设置辐射剂量,进行照射,照射完成后根据实验目的收集细胞或继续培养所需时间。
1.9 临床数据分析
使用TCGA数据库网站cBioPortal(http://www.cbioportal.org/)分析神经营养性酪氨酸激酶受体1(neurotrophic receptor tyrosine kinase 1,NTRK1)基因(TrKA蛋白激酶的编码基因)在卵巢癌病人的遗传变异类型;使用蛋白质组学数据库(https://www.proteinatlas.org/)在蛋白质水平上分析TrKA在正常卵巢组织和卵巢癌组织中的表达情况。
1.10 统计学分析

2 结果
2.1 8种小分子化合物与多腺苷二磷酸核糖聚合酶抑制剂Niraparib在卵巢癌细胞中具有合成致死效应
为了能够找到与PARP抑制剂具有合成致死效应的小分子抑制剂,本文在卵巢癌细胞系SKOV3中使用了PARP抑制剂Niraparib与379种小分子化合物进行联合用药筛选(Fig.1A),这些小分子化合物包括激酶抑制剂、天然产物等。利用CCK8实验将这些小分子化合物与Niraparib进行联合用药筛选,初步筛选所用的Niraparib的药物浓度为2.5 μmol/L, 小分子化合物的药物浓度为10 μmol/L。最终筛选获得8种目标小分子化合物,它们与Niraparib联用对SKOV3细胞的杀伤作用显著高于DMSO处理组和单独使用药物处理组(*P<0.05,**P<0.01)。因此,这8种小分子化合物是与PARP抑制剂具有潜在合成致死效应的小分子化合物(Fig.1B-I)。

Fig.1 Cell viability screens to identify compounds that synergize with Niraparib in SKOV3 cells (A)Screening strategies used to measure synergy of Niraparib with drugs from the small molecular compound library.Plates were seeded(5 000 cells/well)and treated the next day with Niraparib or DMSO.Drugs from the compound library were added on the following day.After SKOV3 cells were treated with compounds(10 μmol/L)and Niraparib(2.5 μmol/L)for 96 hours, the cell viability was analyzed by a CCK-8 kit.Combined with Niraparib, eight compounds(STF-118804(B), Disulfiram(C), GW441756(D), SPHINX31(E), GGTI 298 TFA salt(F), TH302(G), Demethylnobiletin(H), Diosmetin(I))with lethal synthetic effect were screened among 379 small molecule compounds.These results were repeated at least three times.*P<0.05,** P<0.01
为了进一步明确这8种小分子化合物与Niraparib是否具有协同作用,本文通过设计浓度梯度实验,检测不同药物浓度处理后细胞的存活能力,并计算CI值。结果显示,在一定药物浓度范围内,这8种小分子化合物与Niraparib联用能显著降低细胞的存活能力(*P<0.05),并且均具有协同效应(CI<1)(Fig.2A-2P)。其中乙醛脱氢酶抑制剂(disulfiram)(Fig 2A, B)、烟酰胺磷酸核糖转移酶抑制剂(STF-118804)(Fig.2C, D)此前被报道过与PARP抑制剂具有联用效果,佐证了筛选体系的有效性。香叶木素(diosmetin)(Fig.2E, F)和去甲基川陈皮素(demethylnobiletin)(Fig.2G, H)为天然产物,目前的作用靶点尚不明确。GGTI 298 TFA salt(Fig.2I, J)为牛儿基转移酶I(geranylgeranyltransferase I)抑制剂,有文献报道,其可以将细胞周期阻滞在G0/G1期以及促进细胞凋亡[22]。SPHINX31(Fig 2K和2L)为丝氨酸/精氨酸蛋白激酶1(serine/arginine protein kinase 1, SRPK1)的抑制剂。SRPK1磷酸化富含丝氨酸/精氨酸重复序列的剪接因子,是可变剪接的主要调控因子。SRPK1的过表达已在多种癌症类型中被发现,并有助于癌症的发展。Li等人的研究表明,SRPK1通过调节胃癌细胞中的小核仁RNA表达促进肿瘤细胞生长[23]。TH-302(Fig.2M, 2N)为选择性低氧激活的前体药物,靶向实体瘤的低氧区域。但是不幸的是默克制药公司宣布抗肿瘤药TH-302 III期临床试验失败[24]。TrKA(Fig.2O, P)在不同神经元和非神经元细胞的细胞存活、分化和凋亡中发挥重要作用。以往对其他类型实体瘤的研究表明,阻断TrKA可产生抗肿瘤作用[25]。尽管有文献报道TrKA在卵巢癌中也有过表达的情况,但是TrKA在卵巢癌的发生和进展中的作用以及其抑制剂的使用尚未被完全揭示[26],因此,本文选择TrKA的抑制剂作为后续的研究重点。
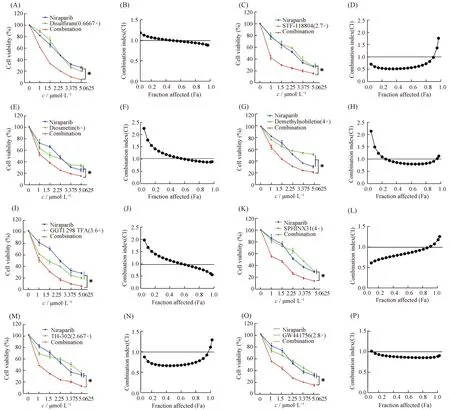
Fig.2 Eight drugs enhanced the chemosensitivity of ovarian cancer cells to Niraparib SKOV3 cells were treated with Niraparib and/or the eight compounds for 96 hours.The cell viability rate was determined by CCK-8 assays.The experiments were repeated three times independently.CI was calculated using CalcuSyn software with the Chou-Talalay equation.CI values reflect the sign and magnitude of drug-drug interaction.CI<1, CI=1, and CI>1 represent synergism, additivity, and antagonism, respectively.These results were repeated at least three times.*P<0.05.The eight drugs, Disulfiram(A-B), STF-118804(C-D), Diosmetin(E-F), Demethylnobiletib(G-H), GGTI 298 TFA salt(I-J), SPHINX31(K-L), TH-302(M-N), GW441756(O-P)and Niraparib have synthetic lethal effects in ovarian cancer cells, respectively
2.2 TrKA在卵巢癌组织中高表达
为了分析NTRK1在卵巢癌病人中的遗传变异情况,本文分析了TCGA数据库中不同机构收集的卵巢癌病人测序数据,结果表明,在所有NTRK1基因的遗传变异类型中,扩增为最多的遗传变异类型(Fig.3A, B)。为了考察TrKA蛋白在卵巢癌组织中的表达情况,本文通过生物数据库分析了TrKA在正常卵巢组织和卵巢癌组织中的表达情况。肿瘤组织及正常组织的免疫组化结果显示,TrKA蛋白在卵巢癌组织中表达增高(Fig.3C, D)。这个结果和前人的免疫组化结果一致[26,27],这就表明TrKA在卵巢癌病人中的过表达是一个潜在的药物靶点。
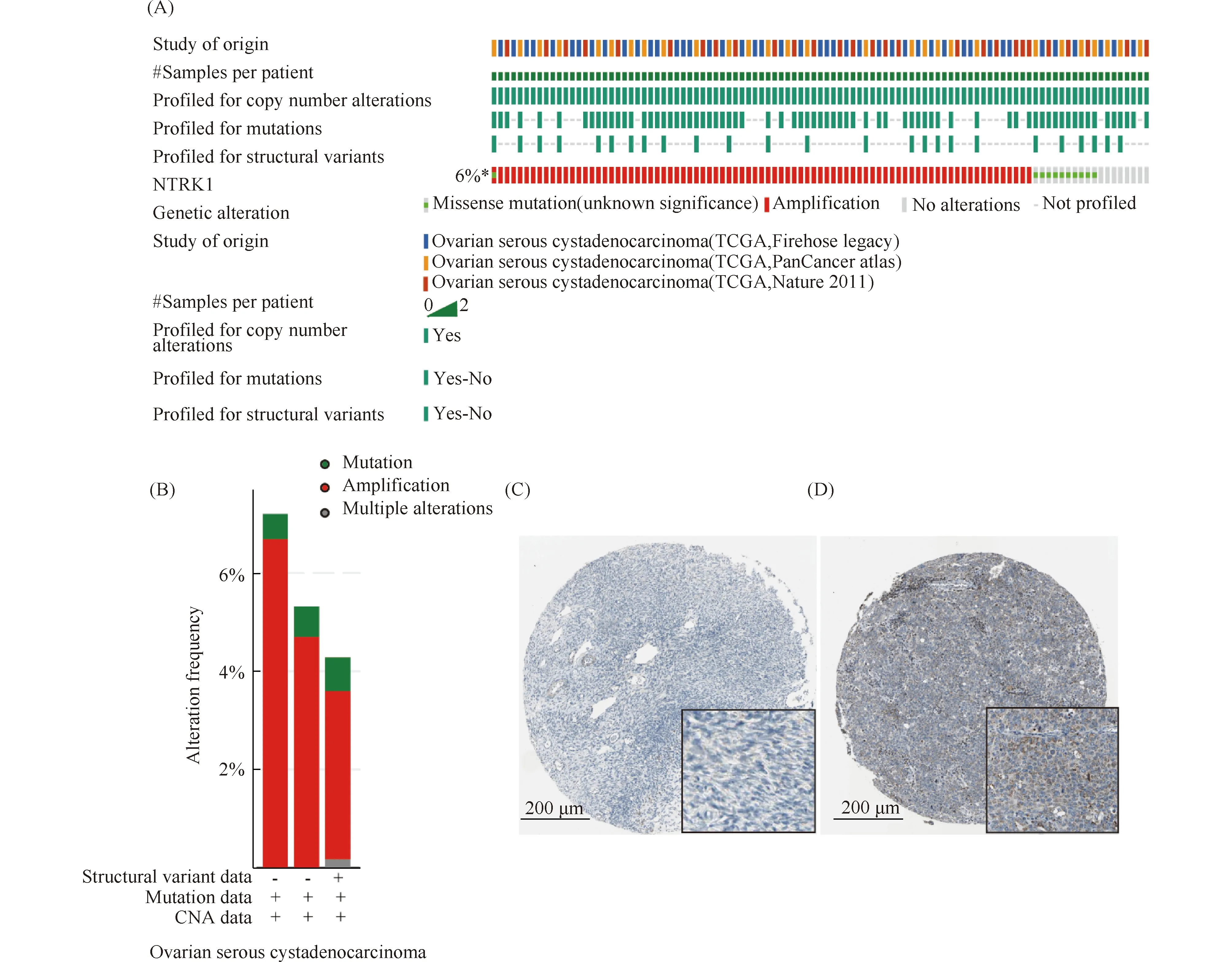
Fig.3 High expression of TrKA in ovarian cancer tissues (A)Analysis of NTRK1 genetic variation in tumor patients by cBioportal database;(B)Sequencing data from various institutional sources indicate that amplification is the most common type of alteration.Representative immunohistochemistry(IHC)images of TrKA proteins for normal ovarian(patient id: 2 344, C)and ovarian cancer(patient id: 2 568, D)samples from the Human Protein Atlas database(https://www.proteinatlas.org/)
2.3 进一步证实TrKA抑制剂与多腺苷二磷酸核糖聚合酶抑制剂具有合成致死效应
为了进一步证实TrKA抑制剂与PARP抑制剂具有合成致死效应,首先通过克隆存活实验观察联合用药的长期效果。结果表明,TrKA抑制剂在1~4 μmol/L的浓度范围内单独用药并不能有效的减少细胞存活,而PARP抑制剂Niraparib对细胞存活的影响具有剂量依赖性。GW441756的药物浓度在1 μmol/L和4 μmol/L时,只要Niraparib的药物浓度大于等于2 μmol/L,2种药物联合使用后,细胞的克隆存活率均显著下降(*P<0.05,**P<0.01,Fig.4A, B)。

Fig.4 TrKA inhibitor and PARP inhibitor had synergistic lethal effects (A)Cells were plated and treated continuously with different concentrations of TrKA inhibitor in combination with Niraparib.After 10-21 days, colonies were stained and clonogenicity was determined.(B)Statistical analysis of colony formation experiments.Data are means ± SD.These results were repeated at least three times.*P<0.05,** P<0.01; Clony formation rates are presented as percentage relative to the control.(C)Inhibition effects of different treatments on cell proliferation was detected by Coomassie bright blue staining
本文也用考马斯亮蓝染色法检测了细胞增殖抑制的情况。将细胞分为4组:即DMSO处理组,单独2.25 μmol/L Niraparib处理组,单独6.3 μmol/L TrKA抑制剂处理组和TrKA抑制剂与Niraparib联合用药组,每隔24 h染色观察细胞的生长情况。相比于DMSO处理组,Niraparib和TrKA抑制剂单独处理可以减慢细胞的增殖能力,而二者联用能够更加显著地抑制细胞的增殖能力(Fig.4C)。综上结果,TrKA抑制剂与PARP抑制剂联合用药具有合成致死效应。
2.4 TrKA抑制剂与多腺苷二磷酸核糖聚合酶抑制剂在其他肿瘤细胞中具有合成致死效应
为了排除由于细胞特异性导致GW441756与Niraparib具有合成致死效应,本文在另外一株卵巢癌细胞系OVCAR-8中进行了验证。结果表明,GW441756与Niraparib在卵巢癌细胞系OVCAR-8中同样具有合成致死效应(**P<0.01,Fig.5A和5B),此结果排除了细胞特异性影响。另外,为了验证是否在其他类型的肿瘤细胞中也有合成致死效应,本文也在人结肠癌细胞系HCT116、人宫颈癌细胞系HeLa和人骨肉瘤细胞系U2OS中进行了验证。结果表明,GW441756与Niraparib联合用药在这些肿瘤细胞中也具有较好的合成致死效应(*P<0.05,Fig.5C-5H)。这就说明,GW441756与Niraparib的联用在肿瘤细胞中可能存在广谱的合成致死效应。
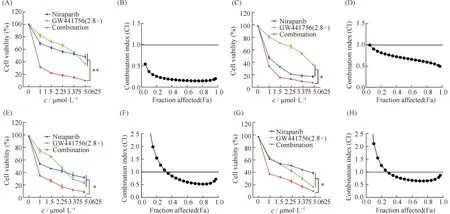
Fig.5 Synergistic effects of PARP and TrKA inhibition are lineage-independent Representative drug response curves of combination therapy with Niraparib and GW441756 for 96 hours in OVCAR-8 cell(A-B), HCT116 cell(C-D), HeLa cell(E-F), and U2OS cell(G-H).CI was calculated using CalcuSyn software with the Chou-Talalay equation.CI values reflect the sign and magnitude of drug-drug interaction.These results were repeated at least three times.*P<0.05,**P<0.01
2.5 TrKA抑制剂不会直接对细胞造成DNA损伤
γH2AX焦点(foci)的多少可以反映DNA损伤断裂程度及修复情况。为明确TrKA抑制剂是否会直接对细胞造成DNA损伤,使用4 μmol/L 的TrKA抑制剂处理SKOV3细胞24 h,免疫荧光染色观察细胞核内γH2AX foci的数目(Fig.6A)。结果表明,DMSO处理组和4 μmol/L的TrKA抑制剂处理组之间的γH2AX foci数目未见显著差异(P>0.05, Fig.6B)。结果表明,TrKA抑制剂本身并不能直接对细胞的DNA造成损伤。
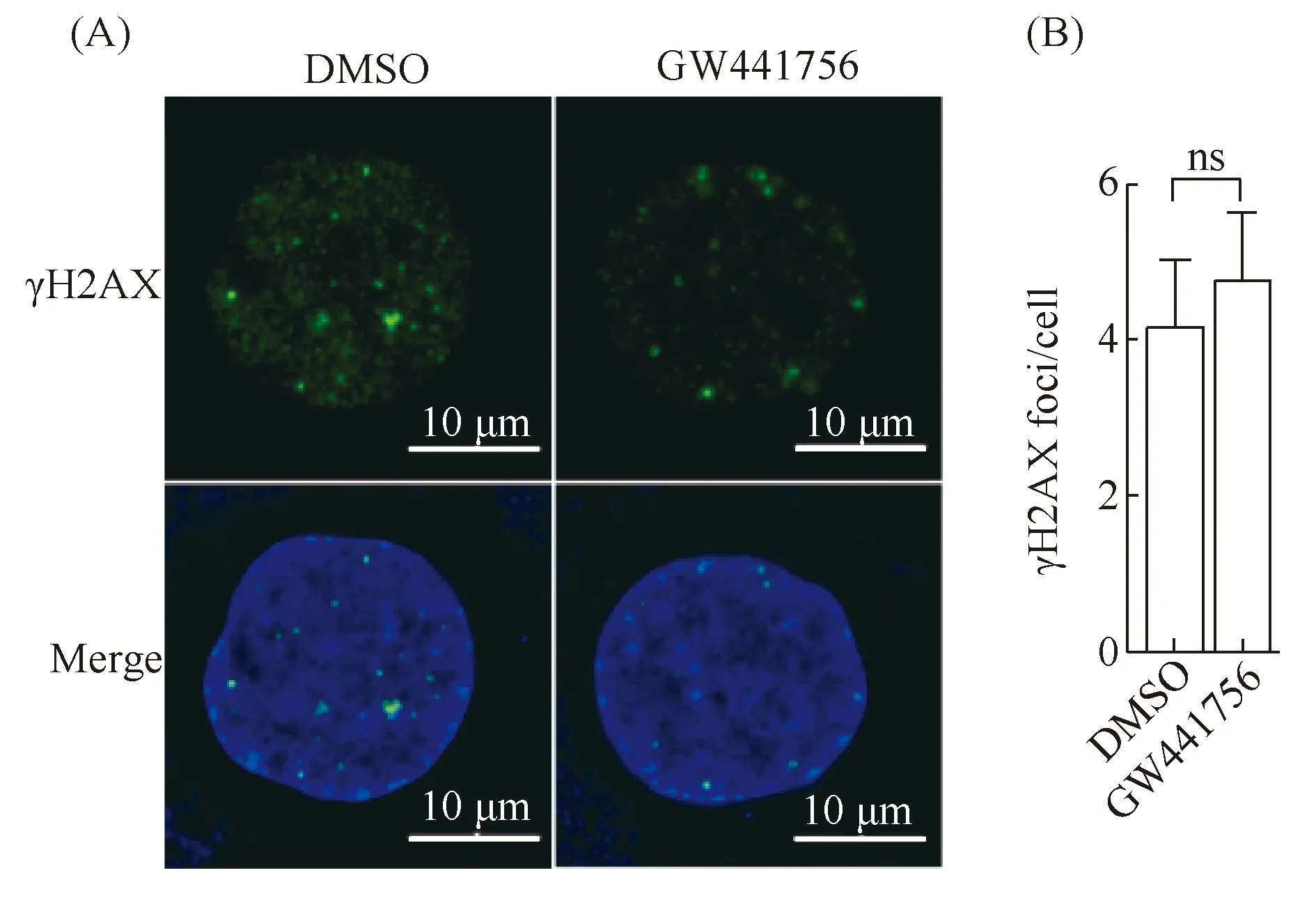
Fig.6 The TrKA inhibitor does not cause DNA damage(A)Immunofluorescence staining analysis of SKOV3 cells for γH2AX(green).DAPI(blue)was used to stain the nuclei.SKOV3 cells were treated with DMSO or GW441756(4 μmol/L)and immunostained with the indicated antibodies at 24 hour post treatment.Scale bar represents 10 μm in all images.(B)The quantified data of γH2AX foci are presented as mean ± SD.These results were repeated at least three times
2.6 TrKA抑制剂阻碍损伤后细胞的DNA损伤修复
为了进一步探究TrKA抑制剂和PARP抑制剂合成致死的机制,首先检测了不同处理后细胞内γH2AX foci的数目变化(Fig.7A)。第1组,单独使用1 μmol/L 的Niraparib处理细胞24 h;第2组,1 μmol/L Niraparib联合4 μmol/L TrKA抑制剂共同处理细胞24 h;第3组,单独使用1 μmol/L Niraparib处理细胞24 h后,换新鲜的培养基继续培养24 h;第四组,1 μmol/L Niraparib联合4 μmol/L TrKA抑制剂共同处理细胞24 h后,换含有TrKA抑制剂的新鲜培养基继续培养24 h。结果显示,第2组与第1组相比,细胞内γH2AX foci的数目未见显著差异(P>0.05),说明TrKA抑制剂未增加PARP抑制剂造成的DNA损伤。第3组与第1组相比,细胞内γH2AX foci的数目显著减少(**P<0.01),说明细胞可以正常修复PARP抑制剂造成的DNA损伤。第4组与第3组相比,细胞内γH2AX foci的数目显著增多(*P<0.05,Fig.7B),说明TrKA抑制剂阻碍了细胞对于PARP抑制剂造成的DNA损伤的修复。

Fig.7 The TrKA inhibitor weakens the DNA damage repair ability of cells after injury (A)SKOV3 cells were treated with 1 μmol/L Niraparib, 3 Gy or 10 Gy of IR, and immunostained with the indicated antibodies at 3 or 24 hours post exposure.(B-C)The quantified data of γH2AX foci are presented as mean ± SD.These results were repeated at least three times.*P<0.05
之后本文分4组进行了电离辐射(ionizing radiation,IR)照射实验:第1组,10 Gy X射线照射细胞,之后释放3 h;第2组,4 μmol/L TrKA抑制剂预处理12 h,再用10 Gy X射线照射细胞,之后释放3 h;第3组,3 Gy X射线照射细胞,之后释放24 h;第4组,4 μmol/L TrKA抑制剂预处理12 h,再用3 Gy X射线照射细胞,之后释放24 h。结果显示,第2组与第1组相比,细胞内γH2AX foci的数目未见显著差异(P>0.05),说明TrKA抑制剂未增加IR造成的DNA损伤。第3组与第1组相比,细胞内γH2AX foci的数目显著减少(**P<0.01),表明细胞可以正常修复3 Gy X射线造成的DNA损伤。第4组与第3组相比,细胞内γH2AX foci的数目显著增多(*P<0.05, Fig.7C),表明TrKA抑制剂阻碍了细胞对于X射线造成的DNA损伤的修复。
本文也使用了碱性彗星电泳实验来观察TrKA抑制剂和PARP抑制剂联用后对细胞DNA损伤修复过程的影响。在SKOV3细胞中,单独使用Niraparib处理24 h或者10 Gy X射线照射后3 h,细胞会产生明显的拖尾现象(Fig.8A),说明Niraparib和X射线对细胞造成了DNA损伤。单独使用Niraparib处理后24 h或者3 Gy X射线照射后24 h,彗星尾部DNA的百分比显著减少(*P<0.01,Fig.8B-8C),表明细胞可以正常进行DNA损伤修复。而在联合处理组,彗星尾部DNA的百分比相比单处理组显著增加(*P<0.05,Fig.8B-8C),证实了TrKA抑制剂会阻碍细胞DNA损伤修复的过程。
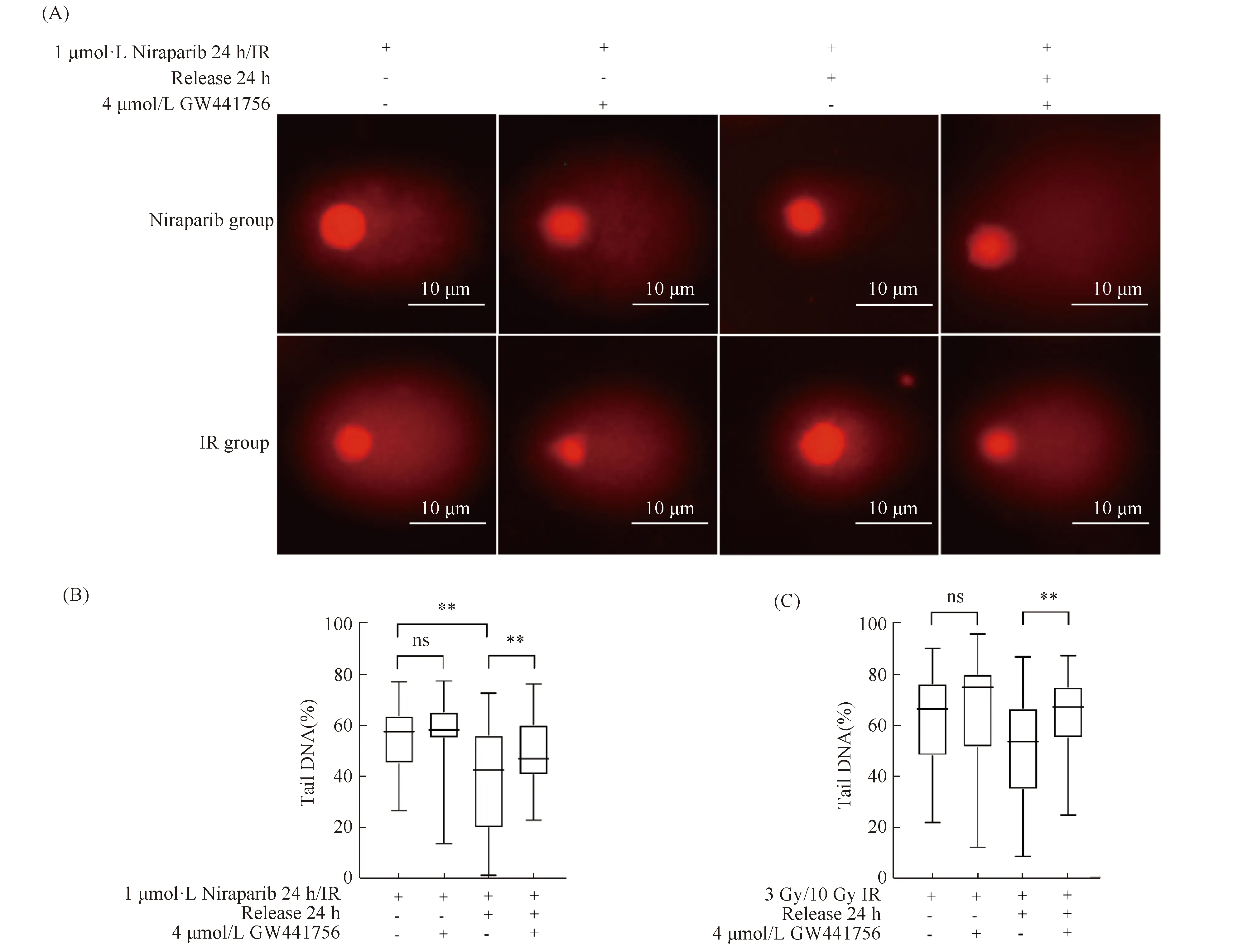
Fig.8 The combination of TrKA inhibitors and PARP inhibitors retards the DNA damage repair ability of damaged cells(A)The levels of DNA damage were examined by comet assays using fluorescence microscopy.(B-C)Box-and-whisker plots of the percentage of DNA in the comet tail from comet assays of SKOV3 cells treated with Niraparib(1 μmol/L), GW441756(4 μmol/L)or IR(3 Gy or 10 Gy).At least 40 cells were analyzed in each sample.These results were repeated at least three times.*P<0.05,**P<0.01
2.7 TrKA抑制剂阻碍细胞的同源重组修复过程
RAD51及53BP1焦点(foci)数目的变化分别是细胞HRR和非同源末端连接(non-homologous DNA end joining,NHEJ)修复效率的评价指标。为了探究TrKA抑制剂是否影响了HRR通路,本文通过免疫荧光检测了不同处理组细胞核内RAD51 foci数目的变化(Fig.9A)。第1组,DMSO处理,细胞核内RAD51 foci的数目较少。第2组,4 μmol/L TrKA抑制剂处理细胞12 h,细胞核内RAD51 foci的数目相比第一组未见显著增多(P>0.05,Fig.9A-B)。第3组,10 Gy X射线照射细胞后释放5 h,细胞核内RAD51 foci的数目显著增加(**P<0.01,Fig.9A-B)。第4组,4 μmol/L TrKA抑制剂预处理12 h,再用10 Gy X射线照射处理细胞后释放5 h,细胞核内RAD51 foci数目相比第2组显著减少(*P<0.05,Fig.9B),表明TrKA抑制剂阻碍了细胞的HR修复。
为了探究TrKA抑制剂是否影响了非同源末端连接修复通路,本文也检测了细胞核内53BP1 foci数目的变化。第一组,DMSO处理,细胞核内53BP1 foci的数目较少。第二组,4 μmol/L TrKA抑制剂处理细胞12 h,细胞核内53BP1 foci的数目相比第一组未见显著增多(Fig.9A-C,P>0.05)。第三组,10 Gy X射线照射细胞后释放2 h,细胞核内53BP1 foci的数目显著增加(**P<0.01,Fig.9A-C)。第四组,4 μmol/L TrKA抑制剂预处理12 h,再用10 Gy X射线照射处理细胞后释放2 h,细胞核内53BP1 foci数目相比第二组无显著差异(Fig.9A-C,P>0.05),表明TrKA抑制剂对细胞的NHEJ修复通路无显著影响。

Fig.9 The TrKA inhibitor impairs the homologous recombination repair process in SKOV3 cells (A)SKOV3 cells were treated with 3 Gy or 10 Gy of IR, and immunostained with the indicated antibodies.RAD51(B)and 53BP1(C)foci were quantified as above.These results were repeated at least three times.*P<0.05,** P<0.01
3 讨论
毫无疑问,PARP抑制剂的出现彻底改变了高级别浆液性卵巢癌的治疗选择,在 BRCA 突变或者HR缺陷的癌症患者中看到了最大的临床益处。尽管PARP抑制剂可以很好地治疗具有HRR缺陷的肿瘤患者,但是越来越多的临床结果表明,耐药性的出现是一个急需解决的科学问题[28]。联合用药是克服耐药性的一个有效途径,目前也已经报道了许多与PARP抑制剂联合用药的成功案例[29]。Lallo等人[30]将PARP抑制剂与Wee1激酶抑制剂结合起来治疗非小细胞肺癌。Niraparib是一种口服选择性PARP抑制剂,也是第一个被批准用于BRCA基因无种系或体细胞突变卵巢癌患者的 PARP 抑制剂[31]。将Niraparib用于一线铂类化疗后的维持治疗以及复发性高级别浆液性卵巢癌的治疗,已经是一个活跃的研究领域。
本文首先从379个小分子化合物中筛选得到了8个潜在的与PARP抑制剂具有合成致死效应的小分子化合物。其中,烟酰胺磷酸核糖转移酶抑制剂(STF-118804)、乙醛脱氢酶抑制剂(disulfiram)与PARP抑制剂的联用已被文献报道。烟酰胺磷酸核糖转移酶可以通过调节组蛋白去乙酰化酶和PARP的活性,从而使肿瘤细胞对药物治疗产生抵抗[32]。PARP抑制剂Olaparib可以诱导溴结构域蛋白4(bromodomain-containing protein 4, BRD4)表达上升,增加乙醛脱氢酶的表达,从而促进NHEJ修复,使细胞产生耐药,因此,乙醛脱氢酶抑制剂与Olaparib可以产生较好的联用效果[33]。
NTRK1扩增作为一种发生在恶性黑色素瘤的致癌事件,与原发肿瘤的侵袭性相关[34]。NTRK是一种非常罕见的靶点,涉及NTRK基因的融合是已知的肿瘤发生驱动因素。针对这些极其罕见的、组成型激活的NTRK 融合的疗法非常有效。尽管 NTRK 融合极为罕见,但是NTRK的其他改变,例如突变、拷贝数改变和转录本的表达增加影响约14%的癌症患者[35]。在NTRK1基因的所有改变中,转录本融合是目前最具特征和最易于药物处理的。DAVIDSON 等人对77例浆液性卵巢癌患者的组织进行免疫组化染色,发现TrKA在晚期卵巢癌中高表达[27]。LAGADEC 等人通过细胞和小鼠的研究发现,NTRK1的过表达增强了乳腺癌细胞的肿瘤发生特性,表明NTRK1可能是乳腺癌治疗的一个潜在靶点[36]。OKAMURA 等人[35]在对11 621例肿瘤病人分析后发现,NTRK1的变异主要包括扩增和mRNA的过表达。Yu等人[37]通过对523例食管鳞状细胞癌病人样本进行荧光原位杂交实验发现,NTRK1基因拷贝数的增加可能是食管鳞状细胞癌患者无病生存期和总体存活的预后指标。我们的研究结果同样表明,TrKA抑制剂与PARP抑制剂的联合使用会产生很好的杀伤肿瘤细胞的作用。TrKA抑制剂虽然不会对细胞造成DNA损伤,但是其会抑制损伤处理后细胞的修复能力。我们在后续的研究中可以通过敲低NTRK1基因或者建立NTRK1的基因敲除细胞系,检测这些细胞是否对Niraparib具有更高的敏感性。此外,也可以利用裸鼠成瘤实验,进一步在小鼠体内研究TrKA抑制剂和Niraparib的联用对肿瘤的杀伤作用,为TrKA抑制剂由基础研究向临床转化提供更多依据。
