四溴双酚A双(2,3-二溴-2-甲基丙基)醚制备工艺技术研究
2022-08-30张成新李远超李秋霞李善清
张成新,刘 扬,李远超,李秋霞,李善清,孟 烨
(山东省海洋化工科学研究院,山东省海洋精细化工重点实验室,山东 潍坊 262737)
多年来,六溴环十二烷(HBCD)一直是建筑外墙保温材料的优选阻燃剂,但由于环保问题,HBCD已在2021年底被禁用。当前,HBCD的替代产品主要有溴化苯乙烯—丁二烯嵌段共聚物(简称“溴化SBS”)和四溴双酚A双(2,3-二溴-2-甲基丙基)醚(简称“甲基八溴醚”)[1-4]。溴化SBS系列阻燃剂制备及应用开发已被美国陶氏化学进行了超前专利保护和技术封锁。近几年,国内相关企业针对甲基八溴醚的制备及应用申请了一些专利保护,但是甲基八溴醚的制备技术仍然存在较多缺陷,缺乏系统的制备技术研究,工业化生产存在诸多问题,比如原料转化率低、副反应多、后处理技术复杂及纯度不高等。根据文献报道,目前甲基八溴醚的制备主要有以下两条技术合成路线:(1)双酚A溴化得到四溴双酚A,然后四溴双酚A与2-甲基-3-氯丙烯(MAC)在碱性条件下醚化得到四溴双酚A双(2-甲基烯丙基)醚(中间体Ⅰ),最后进行烯烃双键的溴化加成制备甲基八溴醚[2]。(2)双酚A与MAC在碱性条件下醚化得到双酚A双(2-甲基烯丙基)醚(中间体Ⅱ),然后进行烯烃双键溴化加成(中间体Ⅲ)和芳环的溴代制备甲基八溴醚[5]。具体合成路线见图1、图2。文章对上述两条技术合成路线进行了详细研究,确定了适合工业化生产的工艺技术合成路线,并对关键工艺参数进行了考察优化,简化了工艺过程,提高了产品纯度和收率,有效降低了原材料成本,可为产业化生产提供有效的技术支持。
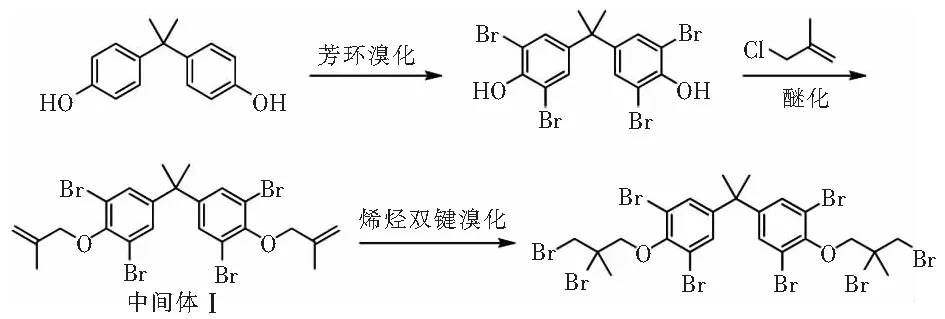
图1 技术路线1Fig.1 Technology roadmap 1

图2 技术路线2Fig.2 Technology roadmap 2
1 实验部分
1.1 仪器与试剂
Bruker Avance 600 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);安捷伦7890A型气相色谱仪;WATERS 2695型高效液相色谱仪;METTLER TGA/DSC1/1100LF热重/差热同步热分析仪;PerkinElmer Clarus 690 SQ8气相色谱—质谱联用仪;PerkinElmer Clarus 690 SQ8液相色谱—质谱联用仪。
双酚A(工业级,韩国LG化学公司);四溴双酚A(工业级,山东天一化学股份有限公司);溴素(工业级,山东天一化学股份有限公司);其余所用试剂均为分析纯。
1.2 合成(技术路线1)
(1)四溴双酚A(TBBPA)的制备[6]。
在1 L反应瓶中依次加入105 g (0.46 mol) 双酚A(BPA),670 g氯苯,搅拌均匀,控制温度在20 ℃~25 ℃,同时滴加140 g(0.943 mol) 溴素与96.2 g 35%双氧水,约2 h滴加完毕,继续保温反应2 h~3 h,然后升温至70 ℃进行强溴化反应30 min。反应结束加入80 ℃的水进行水洗,洗涤后分液,有机层降温至0 ℃~10 ℃进行结晶,过滤、干燥得四溴双酚 A,HPLC纯度99.3%,收率98.3%。
(2)中间体Ⅰ的制备[7-9]。
在1 L反应瓶中加入108.8 g(0.2 mol)四溴双酚A(TBBPA),108.8 g水,机械搅拌。将17.2 g(0.43 mol)氢氧化钠溶于70 g水中,并滴入上述反应体系中。滴加完毕升温至55 ℃~65 ℃反应0.5 h~1 h,体系变成淡黄色清液,然后将39.9 g(0.44 mol)MAC与54.4 g无水乙醇混合均匀后滴入上述反应体系。滴毕,55 ℃~65 ℃反应10 h~12 h。反应完毕,滴加稀酸水溶液调pH值至7,过滤、烘干得中间体I。HPLC纯度98.6 %,收率97.8 %。
(3)甲基八溴醚的制备[8-10]。
在500 mL反应瓶中依次加入65.2 g(0.1 mol)上述反应得到的中间体I、1.63 g催化剂A、228.2 g 1,2-二氯乙烷。20 ℃~30 ℃条件下滴加35.2 g(0.22 mol)液溴,约0.5 h滴毕,继续保温反应1 h,然后升温至35 ℃~40 ℃继续反应20 min,反应结束加入饱和亚硫酸钠水溶液淬灭反应。分液,收集有机相,减压脱溶剂除去1,2-二氯乙烷,加入正庚烷结晶,过滤、烘干得到甲基八溴醚。HPLC纯度97.2%,收率98.8%。m.p.116.7 ℃,1H NMR (600 MHz, CDCl3) δ:7.32(d,4H,J=1.1 Hz), 4.36~4.28(m,4H),4.19(d,2H,J=10.8 Hz),3.99(d,2H,J=10.8 Hz), 2.08(s,6H),1.63(s,6H)。
1.3 合成(技术路线2)
(1)中间体Ⅱ的制备[11]。
在1 L反应瓶中加入45.7 g (0.2 mol)双酚A(BPA),45.7 g水和45.7g无水乙醇,氮气保护,机械搅拌。将24.0 g (0.6 mol)氢氧化钠溶于68 g水中,并滴加至上述反应体系中。滴加完毕升温至50 ℃~60 ℃反应0.5 h~1 h。然后将54.3 g(0.6 mol)MAC滴入上述反应体系,滴毕,55 ℃~65 ℃反应10 h~12 h。反应完毕,滴加稀酸水溶液调pH值至7,脱溶剂除去乙醇和过量的MAC、加入1,2-二氯乙烷萃取,脱溶剂得到中间体Ⅱ,GC纯度89.9 %,收率97.8 %,MS(EI) m/z: 336.0[M+]。
(2)中间体Ⅲ的制备[10]。
在500 mL反应瓶中依次加入50.5 g(0.15 mol)中间体Ⅱ、1.26 g催化剂A、160.0 g 1,2-二氯乙烷。20 ℃~25 ℃条件下滴加50.7 g(0.33 mol)液溴,约1 h滴毕,继续保温反应2 h~3 h,反应结束加入饱和亚硫酸钠水溶液淬灭反应。分液、水洗、收集有机相,减压脱溶剂除去1,2-二氯乙烷,加入乙醇结晶,过滤得到中间体Ⅲ。HPLC纯度98.1%,收率81.3%,m.p. 162.6 ℃,1H NMR(600 MHz,CDCl3)δ:7.18(d,4H,J=8.4 Hz),6.87(d,4H,J=8.4 Hz),4.19(d,4H,J=3.3 Hz),4.16(d,2H,J=10.8 Hz),3.87(d,2H,J=10.3 Hz),1.97(s,6H),1.67(s,6H)。
(3)甲基八溴醚的制备。
在500 mL反应瓶中加入65.6 g(0.1 mol)中间体Ⅲ,1.14 g (0.005 mol)三氯化锑,150.0 g 1,2-二氯乙烷,溶解后置于低温浴槽中降温至0 ℃~5 ℃,滴加67.1 g(0.42 mol)液溴,约1.5 h滴毕,然后升温至25 ℃~30 ℃,保温反应3 h~5 h。反应结束加入饱和亚硫酸钠水溶液淬灭反应。分液收集有机相,减压脱溶剂除去1,2-二氯乙烷,加入正庚烷结晶,过滤、烘干得到甲基八溴醚。HPLC纯度96.2%,收率98.2%。
2 结果与讨论
2.1 中间体Ⅰ的合成条件优化
物料配比即n(TBBPA) ∶n(NaOH) ∶n(MAC)是影响中间体I的收率和纯度的关键因素。在其它工艺技术条件不变情况下,改变TBBPA、NaOH与MAC三者间的配比,实验结果见表1。由表1可知,氢氧化钠用量较少时,中间体I的收率和纯度都较低,主要原因是碱的用量不够,单端醚化杂质较多。通过对氢氧化钠和MAC配比的优化考察,确定最佳的反应料比烯n(TBBPA) ∶n(NaOH) ∶n(MAC)为1 ∶2.15 ∶2.2。
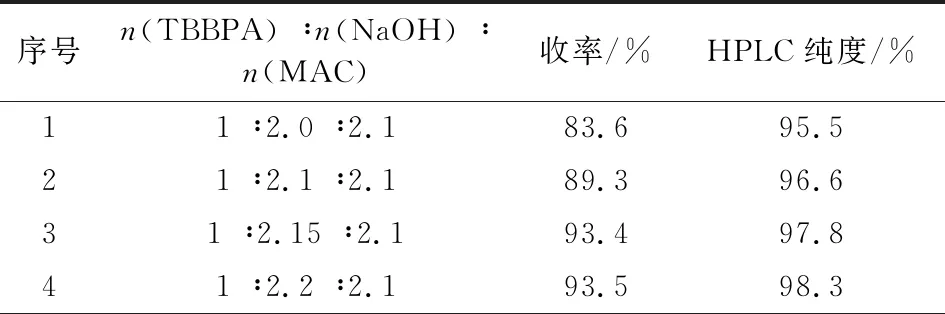
表1 物料比对反应结果的影响Tab.1 The effect of mol ratio on reaction
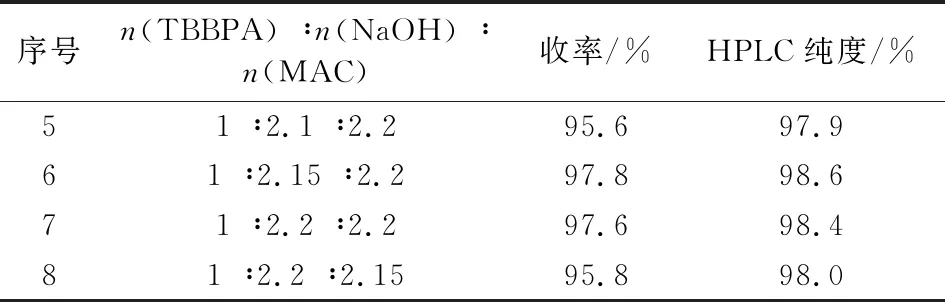
续表1 (Continue)
2.2 中间体Ⅰ的溴化加成条件优化
烯烃双键与溴的加成反应比较容易,适宜的反应条件下可获得较高收率和较好纯度的加成产物。不同的溴化试剂活性差异较大,产物纯度相差较大。四丁基三溴化铵(TBATB)、N-溴代丁二酰亚胺(NBS)为溴化剂时,产物纯度较高;溴素为溴化剂时,产物纯度较差,但在反应体系加入少量催化剂(A、B、C)可明显提高产物纯度,尤其是催化剂A催化时得到的产物纯度可达到97%以上。不同溴化方式得到的甲基八溴醚的热失重(TGA,下同)和溴含量指标不完全相同,从TGA、溴含量、纯度等质量指标和原料成本等综合考虑,选择溴素为溴化试剂,催化剂A为催化剂时原材料成本最低。表2为甲基八溴醚的溴化合成优化条件。
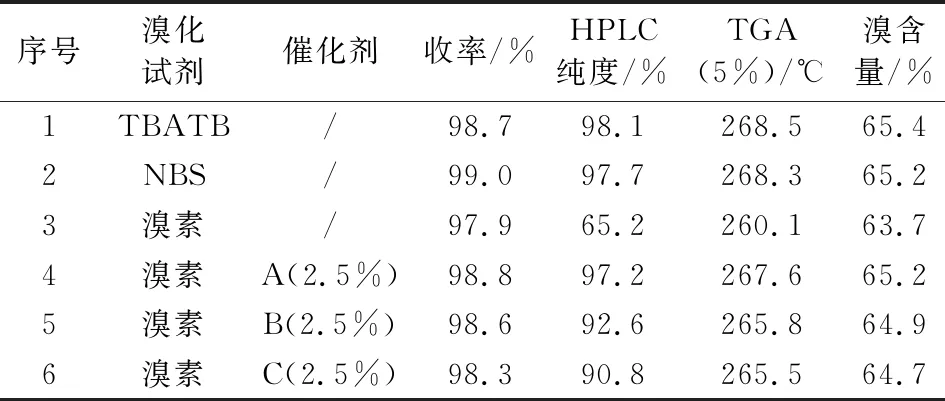
表2 甲基八溴醚的溴化合成条件优化Tab.2 Optimization of the reaction conditions for the synthesis of tetrabromobisphenol A bis(2,3-dibromo-2-methylpropyl) ether
2.3 甲基八溴醚的固化条件优化
甲基八溴醚结构中刚性结构单元(苯环)较少,柔性结构单元(脂肪链)较多,不易形成结晶态,与被阻燃的高分子材料有更好的相容性。由于甲基八溴醚不易形成结晶态,因此较难固化。专利[2]报道使用醇类溶剂或溶有表面活性剂的水为溶剂结晶,通过实验发现,这些结晶方法并不能使甲基八溴醚很好的析出。由于甲基八溴醚极性较小,醇类和水的极性非常大,二者不能互溶,容易形成互不相容的两相,甲基八溴醚易固化成大块固体,给生产造成很大困难。通过对结晶溶剂进行筛选,小极性的溶剂正庚烷做结晶溶剂时,甲基八溴醚可形成细颗粒状晶体析出,且收率较高,结晶溶剂可重复利用。
2.4 中间体Ⅱ的合成条件优化
双酚A比四溴双酚A酸性弱,酚羟基与碱反应生成酚盐能力弱。碱的种类、溶剂、物料配比是影响双酚A醚化的主要因素。从表3可看出,氢氧化钾为碱时醚化反应更好,这主要是氢氧化钾碱性更强,利于双酚A酚盐生成;单独用水为反应溶剂,醚化效果较差,主要原因是水对MAC的溶解性较差,在没有相转移催化剂条件下醚化反应在两相间进行。醇类为溶剂时,醚化效果较佳,甲醇为溶剂时更优,主要是甲醇极性更大,且对氢氧化钾和双酚A的酚盐有较好溶解性,利于醚化反应顺利进行。

表3 中间体Ⅱ的合成条件优化Tab.3 Optimization of the reaction conditions for the synthesis of intermediate Ⅱ
2.5 中间体Ⅲ的溴化条件优化
中间体Ⅲ的芳环溴代相比双酚A的溴代,反应活性要低,需加入适当催化剂来催化反应。不同催化剂对产物纯度影响较大,由表4可知,三氯化锑催化效果较佳,三氯化铝催化效果最差,推测是三氯化铝活性较高,产生了多溴代杂质。
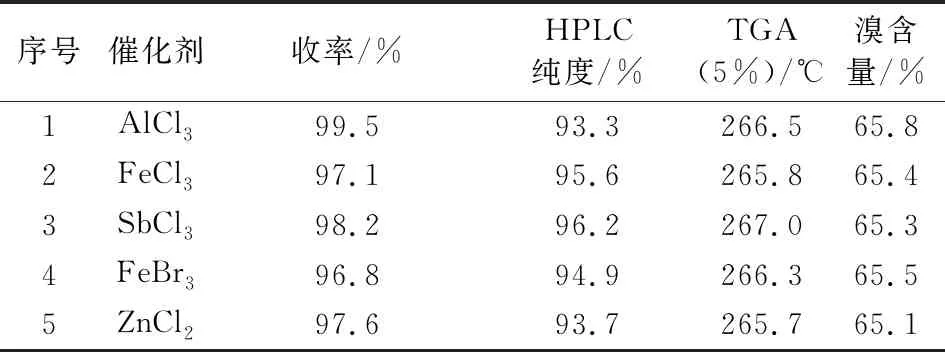
表4 甲基八溴醚的合成条件优化Tab.4 Optimization of the reaction conditions for the synthesis of tetrabromobisphenol A bis (2,3-dibromo-2-methylpropyl) ether
2.6 不同技术路线获得的甲基八溴醚产品指标的对比
技术路线1的收率较高,产物纯度、溴含量、色度等指标较佳。工艺路线2的收率相对较低,双酚A的醚化过程存在单端醚化杂质较多的问题,中间体III的溴化需要加入催化剂才能较好的实现溴化,产物纯度相对较低一些。指标对比见表5。
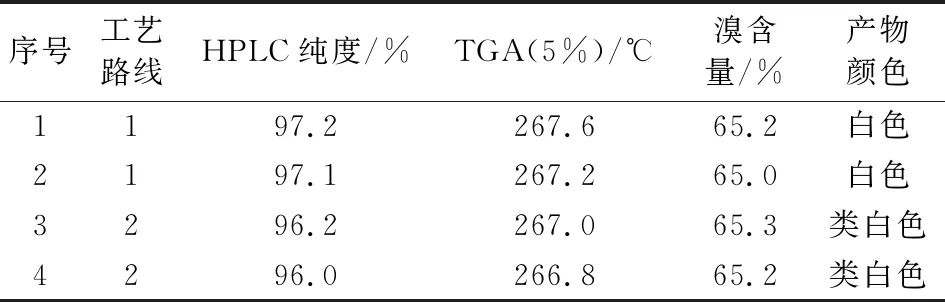
表5 甲基八溴醚的技术指标对比Tab.5 Comparison of technical indicators of tetrabromobisphenol A bis(2,3-dibromo-2-methylpropyl) ether
3 结论
文章对甲基八溴醚的两条制备路线进行研究,通过对比可发现,技术路线2工艺难度大、收率偏低、产品纯度和色度不佳。相比而言,技术路线1更易实现,收率更高,产品纯度更好,且四溴双酚A已有工业化产品,以四溴双酚A为原料进行甲基八溴醚的制备,产业化配套更容易实现,成本更低,更适合工业化生产。
