过渡金属双功能催化剂在碱性条件下电解水
2022-08-25康建光孙梅娟
田 臻,康建光,孙梅娟,王 飞,张 静
(太原科技大学化学与生物工程学院,山西 太原 030024)
电化学水分解被认为是氢生产的最有前途的技术之一。这一能源的开发和利用可实现以“水”、“氢气”和“氧气”为循环的能量转换体系,实现真正意义上的“零排放”。电解水的主要挑战是使用高效电催化剂,降低两个半反应的高过电位并加快反应速率[1-2]。目前,贵金属Pt和 Ir/Ru基催化剂被认为是催化HER和OER最有效的电催化剂。然而由于其储量低、成本高,在商业应用中受到很大限制[3]。因此,为了替代这些昂贵的催化剂,研究人员致力于探索和开发具有低成本、高活性以及良好稳定性的非贵金属电催化剂,从而使整体水分解反应更切实可行。特别地,在同一电解质中制备具有高活性,大电流密度以及优异耐久性的双功能电催化剂仍然是目前所面临的主要挑战[4]。本文综述总结双功能水分解电催化剂的最新进展,探讨水电解的基本原理,然后描述双功能催化剂的设计概念及其电化学性能,最后提出未来研究方向的挑战和展望,以期能为设计和开发电解水双功能催化剂提供更多的新思路。
1 电解水反应机理
电催化水分解是利用催化剂将电能转化为化学能的过程,在标准条件下吉布斯自由能(ΔG)=237.2 J·mol-1,水分解反应吸热性强,标准条件下驱动电化学水分解需要 1.23 V的电压。
1.1 电解水析氧反应
氧气析出反应(OER)是电解水制氢的阳极反应,也是ORR的逆反应过程。由于OER 是四电子反应过程,且O—O 键的形成需克服较高的化学反应能垒,导致OER反应的动力学很缓慢[5]。因此,高效稳定的OER电催化剂的开发对提高整体水分解性能十分重要。碱性条件下OER的机理如下所示。
(1)OER过程的初始步骤,OH-在催化剂表面吸附,形成M-OH中间体。
M+OH-→M-OH+e-
(1)
(2)M-OH与OH-进一步结合生成与H2O和电子耦合的M-O中间体。此后,O2可以通过两种不同的途径生成。一种是两个M-O直接结合产生O2和M活性位点。
M-OH+OH-→M-O+H2O+e-
(2)
2M-O→2M+O2
(3)
另一种是通过M-O与OH-耦合生成M-OOH中间体,然后继续与OH-结合产生O2和活性位点。
M-O+OH-→M-OOH+e-
(4)
M-OOH+OH-→M+O2+H2O+e-
(5)
在酸性电解质中 OER 的四电子转移步骤类似于碱性条件:
M+H2O→M-OH+H++e-
(6)
M-OH→M-O+H++e-
(7)
2M-O→2M+O2
(8)
M-O+H2O→M-OOH+H++e-
(9)
M-OOH→M+O2+H++e-
(10)
由于上述反应存在多种中间体,很难完全确定 OER 的速率决定步骤并对 OER 的动力学进行分析。此外,在酸性介质中,Ru/Ir 的氧化物是OER 电催化剂的最佳选择,而大多数非贵金属电催化剂由于在酸性电解质中易腐蚀,稳定性较低,因此,过渡金属衍生的电催化剂在碱性条件下更有利于催化OER。
1.2 电解水析氢反应
氢气析出反应(HER)发生在电解水阴极,是典型的两电子途径。HER 的速率高度依赖于电解质的 pH,在不同的电解质中,HER 电催化机理也不相同[6]。理论研究表明,酸性电解质中的 HER 活性与氢吸附(Hads)有关,由 Volmer/Heyrovsky 或 Volmer/Tafel 步骤组成,如下所示:
(1)氢离子与催化剂(M)表面活性中心上的电子反应,形成吸附氢 (MHads)。
Volmer 反应:H3O++M+e-→MHads+H2O
(11)
(2)水合氢离子伴随一个电子,扩散到 MHads生成氢分子。
Heyrosky 反应:H3O++MHads+e-→H2+H2O
(12)
(3)或者两个 MHads在电催化剂表面上扩散并反应生成 H2。
Tafel 反应:2MHads→H2+2M
(13)
在碱性电解质中:
(1)由于缺少 H+,H2O 分子代替 H+与电子偶合,转化为吸附的OH-和吸附在电催化剂上的氢原子(MHads)。
Volmer反应:H2O+M+e-→OH-+MHads
(14)
(2)H2O 分子与 MHads得到一个电子生成氢分子。
Heyrosky反应:H2O+MHads+e-→OH-+H2+M
(15)
在碱性介质中吸附氢前需要H—O—H键的断裂,比在酸性条件下还原 H3O+更难实现。基于Volmer 和Heyrosky 的反应途径,活性位点上的水吸附、水解离能力、氢结合能和水溶液中OH-的吸附,这些因素对碱性条件下的HER 性能影响较大。目前,研究者广泛开发高活性HER电催化剂,使其能够提高 HER性能,如过渡金属硒化物、氮化物、磷化物和碳化物[7]。
1.3 双功能电催化剂
电催化剂在水分解中的双功能性是指催化剂可以同时进行氧化还原反应(水还原和氧化)的能力。图 1 展示了具有双功能催化剂的单电池电解水示意图。
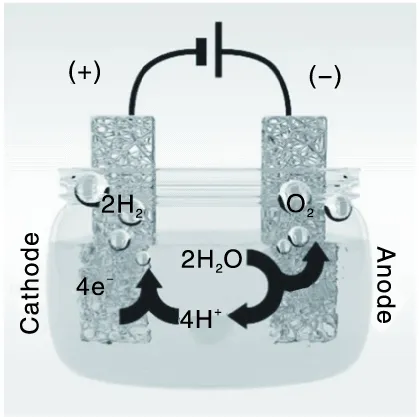
图1 电解水装置示意图Figure 1 Water electrolysis on single electrode with bifunctional catalyst
目前,已经开发出大量低成本和性能优异的HER 或OER 电催化剂,但在实际条件下,将两个电极反应集成在同一电解质中仍然存在挑战。值得注意的是,探索一种双功能电催化剂可以同时有效催化 HER和OER,有利于简化电解槽的设计,避免不同电催化剂在两个电极上的交叉效应,并大大降低器件的制造和运行成本[8]。因此,开发自支撑双功能电催化以促进整体水分解过程至关重要,进而应用于大规模的商业化电解水。
2 过渡金属基双功能电催化剂
2.1 过渡金属氧化物
由于低成本、高活性和优异的稳定性,过渡金属基氧化物(TMO)为开发具有高效电催化性能的双功能催化剂提供了很大的可能性。不同物质之间产生的协同效应改变了催化剂的电子结构,变化的电子结构反过来影响电催化的整体性能。目前,异质结构和杂化化合物之间的纳米界面引起了广泛关注。如Qayoommugheri A等[9]通过化学生长法偶联 Co3O4/NiO 纳米结构,金属氧化物之间的协同效应增强了材料的导电性,复合材料的界面化学增加了水的解离和中间体的吸附,从而降低了HER和OER 的过电位。Liu J等[10]利用 Co3O4@MoS2互利的异质结构改善缓慢的动力学,在 1 mol·L-1的KOH溶液中,当电流密度为 10 mA·cm-2时,HER和OER的过电位分别为 90 mV和 269 mV,相应的 Tafel 斜率分别为 59.5 mV·dec-1和 58 mV·dec-1。在全水解过程中,电池电压仅为 1.59 V 时电流密度可达到10 mA·cm-2。
将催化剂与导电基底结合是提升催化剂催化活性的常用方法。其中具有高导电性的多孔结构碳材料是目前使用较多的导电基底。Liu B等[11]通过在二维(2D)黑色磷(BP)上生长非晶态过渡金属(钴和铁)氧化物,制备了一种双功能电催化剂(CoFeO@BP)。使用 CoFeO@BP 作为电解槽的阳极和阴极,只需 1.58 V 的电池电压即可在 1 mol·L-1的KOH中达到 10 mA·cm-2的电流密度,且 HER和 OER过电位分别为 88 mV和266 mV。Zhou Qianqian等[12]制备了具有核壳结构的CuOx@Co3O4NRs/CF,由于其大的电化学表面积以及 CuOx核和 Co3O4壳之间的协同效应,CuOx@Co3O4NRs/CF可以连续产出 O2或 H2至少 24 h,并且 OER 和 HER 的法拉第效率分别达到 99.7% 和 96.4%。二元金属 NiFe 基催化剂由于丰富的催化位点和 NiFe 固有的催化活性,在 OER、HER 和整体水分解方面表现出优异的电催化性能。Lin Y等[13]制备了 NiFe-MnCo2O4/NFF 催化剂,3D花状结构的MnCo2O4和NiFe纳米粒子的高导电性协同增强电子转移并减小整个水分解过程中的扩散距离,在催化过程中促进了电子的传导。
2.2 过渡金属氢氧化物
过渡金属氢氧化物被认为具有很高的OER电催化特性,并且也适用于催化 HER。通过水热-硒化-水热的方法将NiFe层状双氢氧化物(LDH)生长在垂直排列在泡沫镍(NF)上的CoSe纳米管表面(CoSe@NiFe-LDH/NF)[14],由于独特的纳米结构,提升了界面处含氧物质的吸附/解吸能力,在1 mol·L-1的KOH 溶液中,全水解时仅需1.53 V 的电池电压即可达到 10 mA·cm-2的电流密度,此时OER和HER的过电位分别为 201 mV 和 98 mV。优于最先进的商业贵金属基电催化剂。Liang X课题组[15]制备的新型无粘合剂 NiFeLDH@Ni3S2异质结构双功能催化剂仅需1.74 V 的低操作电压即可达到20 mA·cm-2的水分解电流,可实现稳定循环的整体水分解。过渡金属基材料中引入杂原子掺杂为提高电化学性能开辟了一条非常有效的途径。如Fan R等[16]通过使用溶剂热法将原子 Ir 掺杂到 NiCo 层状双氢氧化物 (LDH),得到一种双功能催化剂Ir-NiCo LDH,在电流密度为 10 mA·cm-2时 HER和OER的过电位分别为 21 mV和 192 mV。全水解时仅需 1.45 V的电压,稳定性达 200 h。
目前,已经报道了几种制备高效催化剂的有效策略,主要包括催化剂结构的设计以及优良导电材料的耦合,进一步提高电催化剂在实际电解水中的功能活性。Yu L课题组[17]制备了具有核壳纳米结构的高效 3D 电催化剂,其中几层 NiFe LDH 纳米片生长在由 Cu 泡沫支撑的 Cu 纳米线核上,以实现整体的水分解。值得注意的是,Mn+1XnTx(n=1~3,M是过渡金属,x 是 C 和/或 N,T 为化学基团,如-OH、-O、-Cl 和 -F) 已经在广阔的领域内激发研究者兴趣。它们具有与费米能级附近的态密度相关的高电导率、优异的亲水性和通过接枝的化学基团实现丰富的表面化学等优点。因此,3D MX 框架不仅可用于促进催化剂上的质量/电荷传输,而且还通过增强催化剂上的水吸附/活化来加速 NiFe-LDHs 的 OER 氧化还原过程和 HER 的 Volmer 步骤[18]。
2.3 过渡金属磷化物
受益于高导电性、金属特性以及 P 位点的电负性可以捕获质子的特性,过渡金属磷化物 (TMPs) 可作为贵金属和氧化物基电催化剂的优质替代品,用于整体水电解[19]。目前,已通过磷化、金属掺杂和理论计算等策略开发了大量过渡金属磷化物电催化剂。研究发现稀土 (Er) 元素掺杂的 CoP 与纯 CoP 纳米片相比,Er 掺杂的 CoP 超薄纳米网 (NMs) 具有更大的比表面积 (123.4 m2·g-1) 和双层电容 (143.1 mF·cm-2),因此具有更多的活性位点[20]。双电极电解系统在碱性介质中仅需1.58 V 的电压即可驱动 10 mA·cm-2的电流密度,并能够保持稳定电解 25 h。密度泛函理论(DFT)计算表明,催化剂的优异性能源于较小的过电位和氢吸附能。由于异质双金属磷化物在结构和化学方面的优势,其在双功能电催化剂领域内表现出更高的电催化活性。Li J等[21]通过水热和磷化合成了生长在 3D 泡沫镍上 NiCoP 纳米线 (NiCoPNWAs/NF),在 1.0 mol·L-1的KOH 中,电流密度为 100 mA·cm-2时,HER 和 OER 的过电位分别为 197 mV和370 mV。由于多孔结构可以提供良好的表面润湿性和充分暴露的活性位点,磷酸盐对磷化物的电化学性能具有良好的催化性能[22],通过磷化 Ni-Co 层状双氢氧化物(Ni-Co LDH)制备得到的 NCPP NCs双功能电催化剂,在 1.0 mol·L-1的KOH 中,OER 和 HER 仅需要 291 mV 和 140 mV 的过电位即可实现 10 mA·cm-2的电流密度。此外 Prashanth W Menezes等[23]制备了一种具有显著结构特征的磷基无机材料亚磷酸镍 Ni11(HPO3)8(OH)6,在碱性介质中具有极高的结构稳定性和耐久性。Zhang W等[24]通过阳离子空位工程成功地优化了 Ni2P,获得 V-Ni2P/NF 双功能催化剂。由 V-Ni2P/NF电极作为水电解装置的阳极和阴极,仅需 1.59 V 的电压,电流密度即可达到 10 mA·cm-2,并且在 50 h 内无明显衰减。
2.4 过渡金属氮化物
过渡金属基氮化物(TMNs)被认为是水电解的良好催化剂,在过渡金属中引入氮,通过增加费米能级附近的态密度(DOS)来改变金属的电子结构,而DOS 的重新分布又会产生类似于贵金属的电催化性能。因此,在电化学催化反应中,金属氮化物比相应的金属具有更高的电催化活性[25]。由于单相 TMNs 表面的 M-H 结合能大于 Pt 基催化剂,因此催化活性仍然比 Pt 基金属差,另外,一些单相氮化物的稳定性较差,通常引入另一种金属构成双金属氮化物或与其他材料进行复合来调控催化剂表面电子结构,从而优化其 M-H 结合能,实现催化活性的提升。如Wang Y课题组[26]通过 LDH 的简单氮化反应合成了一种由纳米颗粒堆积的多孔 Ni3FeN 电催化剂(NSP-Ni3FeN),凭借独特的结构优势,NSP-Ni3FeN/NF 表现出优异的电催化性能,在 10 mA·cm-2下 HER 和 OER 具有45 mV和 223 mV 的极低过电位,全水解时仅1.495V 的电压便可驱动 10 mA·cm-2的电流密度。另外,高度多孔的金属氮化物的形成可以显著提高活性表面积进一步暴露更多的活性位点,促进离子扩散和电子转移[27]。通过简单的电沉积以及在 NH3气氛中进行退火,在多孔泡沫镍上合成镍-钴氮化物(NiCo2N)纳米片,使用该电极作为水分解系统的阴极和阳极时,具有高产气率和良好的稳定性。Yin Z等[28]制备了一种双金属 Ni-Mo 氮化物纳米管电催化剂。由于催化剂的内部空隙和多孔壁的结构保证了电解质与催化剂的良好接触,并提供了大量暴露的活性位点,从而有助于催化活性的提高。在碱性水电解槽中仅需 1.596 V 的电压即可实现 10 mA·cm-2的电流密度。原子层沉积(ALD)作为纳米级材料的精细合成技术,在制备或改善电催化剂活性等方面具有很大优越性。Guo D等[29]通过静电纺丝和原子层沉积制备了SFCNF/Co1-xS@CoN,该复合材料在酸性和碱性溶液中均表现出很强的电催化活性,是贵金属基电催化剂的有效替代品。
2.5 过渡金属硫化物
过渡金属硫化物 (TMS) 具有独特的结构特征、丰富的活性位点、可调节的电子性质和成分,是研究 HER 和 OER 的双功能电催化剂之一[30]。亲水性介质和前体可以用于合成亲水性电催化剂,用于整体水分解。硫化钴 (Co3S4) 催化剂表现出具有亲水表面的层状纳米结构,可以促进水基底物向电极孔隙和活性位点的扩散[31]。在电解槽中电解水时 Co3S4表现出比 Pt/C-RuO2催化剂更好的性能和稳定性。Wang C等[32]通过溶剂热法制备了生长在泡沫镍上的 Ni3S2和晶态 MoSx纳米片(Ni3S2-MoSx/NF),作为一种高效的催化剂用于整体水分解。由于 Mo-S-Ni 界面增强了析氢和含氧中间体的化学吸附,NiMoS 催化剂在 10 mA·cm-2下对 HER 和 OER 分别表现出 78 mV 和 260 mV 的低过电位,优于商业基准(IrO2和Pt/C),使用 NiMoS 作为双功能催化剂组装的双电极碱性电解槽可以在 1.53 V的低电压下驱动 10 mA·cm-2的电流密度,并具有超过 100 h的卓越稳定性。二维纳米阵列核壳结构可以在异质界面提供更多的活性位点,促进气泡释放。Yang Y等[33]制备了自支撑且富含异质界面的 Ni3S2@FeNi2S4@NF 电催化剂。密度泛函理论计算表明,Ni3S2和 FeNi2S4之间形成的异质界面可以优化 H*吸附的吉布斯自由能 (DGH*),使其显示出优异的电催化活性。
元素掺杂(如S、Fe、Sn)可以有效地改变 TMS 的电子结构并调整电子密度以调节 H*和 O*、OOH*吸附自由能,以获得高 HER 和 OER 性能,Jian J课题组[34]报道了通过简单的水热法在泡沫镍上生长 Sn 掺杂的 Ni3S2纳米片(Sn-Ni3S2/NF),Sn4+的掺杂增加了有效的活性位点以及增强了催化剂本身内在活性,水解过程中,当电流密度为 10 mA·cm-2时,所需电压仅为 1.46 V。同样,通过水热法在泡沫镍上制备铁掺杂的 Ni3S2纳米线,在进行全水解时仅需 1.95 V电压即可达到 500 mA·cm-2的高电流密度,且持续时间超过 14 h[35]。
2.6 大电流
大规模电催化全水解过程中,在高电流密度下会迅速形成大量的 H2/O2气泡。气泡聚集在电催化剂和电解质的接触面上,严重阻碍了液体的质量传输,减缓了电子传输,减少了暴露的活性位点数量,导致电催化的活性和稳定性降低[36]。研究发现,电催化剂原位生长在导电基材上,避免了粘合剂和导电剂的使用,简化了电极的制作工艺,降低了成本。底物和电催化剂的紧密结合,不需要额外的粘合剂,确保电荷快速转移,同时紧密的整合有效防止了电催化剂的脱落。在碱性电解液中的大电流电催化剂,几乎都是自支撑过渡金属基电催化剂。如Jian J等[37]通过水热法制备了原位生长在泡沫镍上的 K2Fe4O7纳米晶体催化剂,被用作电解槽的阴极和阳极时,该水电解槽可以在 1500 mA·cm-2的电流密度下稳定运行超过 60 h,在 1.0 mol·L-1的KOH 溶液中的电压为 2.01 V,在 10.0 mol·L-1的KOH 溶液中为 1.89 V,体现了这种双功能电催化剂的优异催化性能。Qian G等[38]在 3D 泡沫镍上构建自支撑的超薄 N 掺杂石墨烯封装的 Ni 纳米粒子与 MoO2纳米片,可以在 1000 mA·cm-2下电解超过 196 h,而阴极和阳极活性没有明显下降,表明具有优异的耐用性。
异质结构电催化剂的构建可以充分发挥各组分的优势用于整体水分离,尤其是在大电流密度下运行,但能稳定发挥的复合物数量相对较少。Zhai P等[39]制备了分层过渡双金属氧化物/硫化物异质结构(NiMoOx/NiMoS/NF)阵列催化剂。组装为双电极电池,1.0 mol·L-1的KOH 溶液中,在 500 mA·cm-2和 1 000 mA·cm-2的高电流密度下,电池电压仅达到 1.60 V和 1.66 V,并且能在 500 mA·cm-2的高电流密度下连续工作 500 h,具有良好的长期稳定性。Kou T等[40]制备了一种 3D PNi 电催化剂,在 1.0 mol·L-1的 KOH 电解质中,该催化剂可以实现 1 000 mA·cm-2的高电流密度,HER 和 OER 过电位分别为 245 mV 和 425 mV。经过理论计算研究了界面诱导的协同作用,表明 RuO2-NiO 界面可以有效地调整电子结构,增加电导率,优化中间体结合,促进 H2O 解离,从而有利于 OER 和 HER 过程。此外,RuO2-NiO/NF 催化剂还表现出高稳定性,可以在 1 000 mA·cm-2的电流密度下连续电解 2 000 h[41]。表1总结了催化剂相应的 HER 和 OER 电化学性能。
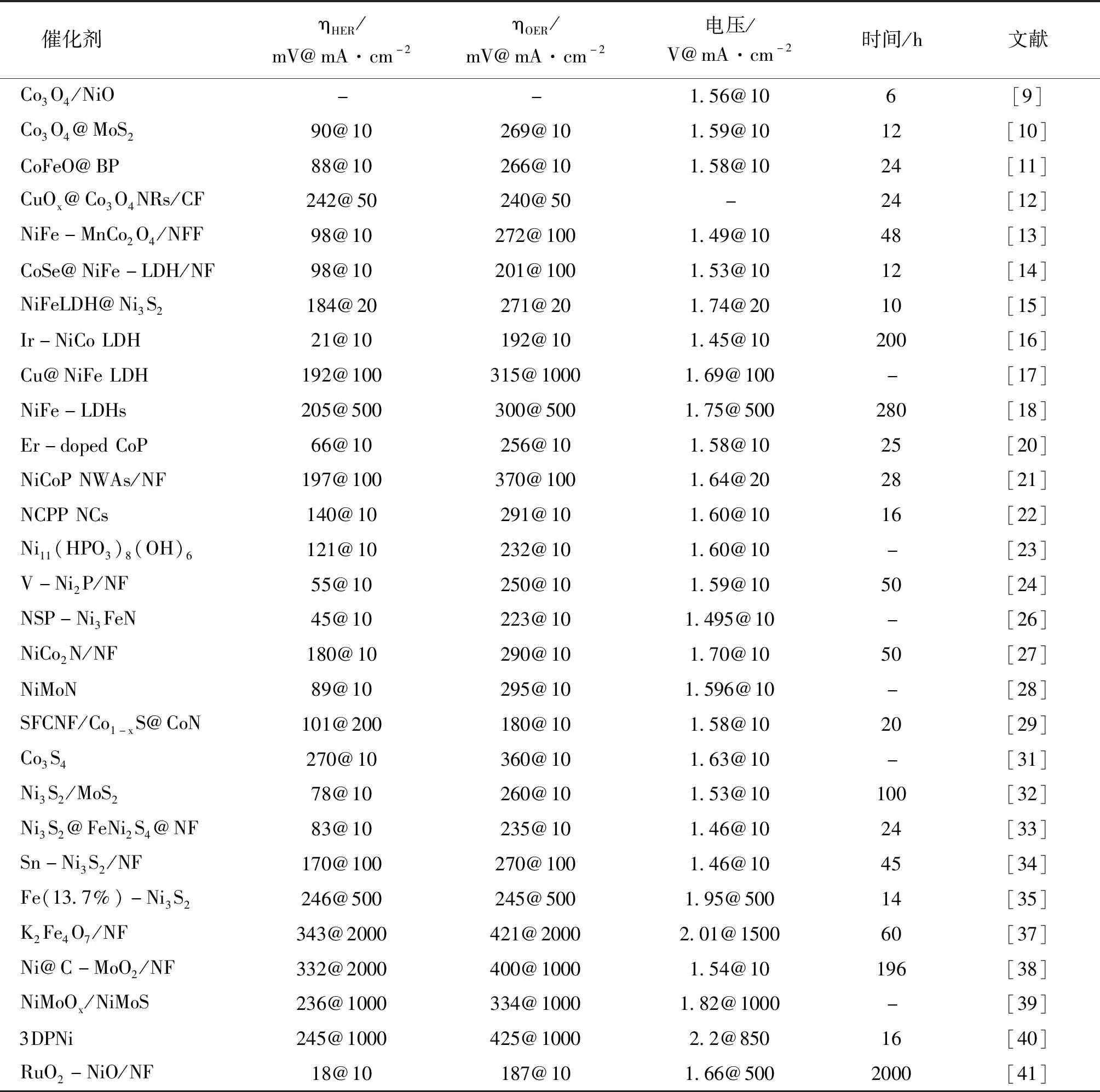
表1 双功能催化剂电化学性能总结Table 1 Electrochemical performance of bifunctional catalysts HER and OER
3 结 语
随着对可再生和可持续能源资源的不断探索,开发双功能电催化剂用于整体水电解,成为未来生产清洁氢气的有效方法。概括了在碱性条件下,用于整体水分解的非贵金属双功能电催化剂的最新进展,包括过渡金属氧化物、磷化物、硫化物、氢氧化物、氮化物以及在大电流条件下的非贵金属催化剂。尽管目前在制备双功能电催化剂方面取得了巨大的进步和许多成就,但仍面临一些尚未解决的问题,需要进一步的研究。(1)由于大多数过渡金属基 OER 电催化剂在低 pH 值电解质中的稳定性较差,因此,在较宽 pH 范围内起作用的双功能电催化剂的开发仍处于初始阶段;(2)水分解的两个半反应 HER 和 OER 具有不同的活性位点,这使得设计一个对两个反应都有活性的单一电催化剂成为挑战,需要对电催化剂的设计策略进行进一步改进;(3)除了实现双功能催化外,在大电流密度下能够稳定运行的电催化剂也是商业电解槽需要攻克的另一难题。
