鱼类环境DNA metabarcoding片段的近缘物种识别差异
2022-08-17陈治马春来叶乐杨超杰王海山
陈治 ,马春来 ,叶乐 ,杨超杰 ,王海山 *
( 1.海南热带海洋学院 热带海洋生物资源利用与保护教育部重点实验室,海南 三亚 572022;2.海南热带海洋学院 海南省热带海洋渔业资源保护与利用重点实验室,海南 三亚 572022)
1 引言
近年来,环境 DNA(Environmental DNA,eDNA)metabarcoding技术开始广泛应用于渔业生态学领域,在物种多样性调查、外来鱼种监测、濒危鱼类保护方面展现了极大的应用价值和发展潜力[1-2]。其中,metabarcoding片段的选择对该技术的有效使用至关重要[2-3]。经过众多研究者的不懈努力,目前已筛选出一些物种识别率较高、对应引物扩增能力较强的优质片段[3]。如Miya等[4]基于880种硬骨鱼类线粒体12S序列设计的MiFish-U,扩增片段长度控制在163~185 bp范围内,对冲绳水族馆的鱼类检出数目高达230种;Vences等[5]针对线粒体16S设计的脊椎动物Vert-16S,不仅扩增片段长度更长(约260 bp),而且对鱼类、两栖类均具有较好的适用性—以德国境内的两条河流为验证水域时,物种检出率高达82%~93%;Balasingham等[6]设计的PS1,将扩增片段选定于线粒体COI基因上,在获得较多鱼类可操作分类单元(Operational Taxonomic Units,OTUs)的同时,可高效利用公共数据库内的参考序列进行物种注释。截至2020年,被报道的鱼类eDNA metabarcoding片段已多达数十个,其中表现优异的片段约10余个[3]。各片段的筛选背景不同,实际表现也各具特色,这为丰富鱼类eDNA metabarcoding库、推动鱼类多样性研究奠定了坚实基础。
然而,迄今为止的鱼类eDNA研究尚不健全,如何根据研究水域和目标类群合理选择metabarcoding片段仍处于探索阶段[3]。其中,metabarcoding片段对近缘物种的适用性就是一个亟需解决的问题。物种识别率和引物通用性是选择鱼类eDNA metabarcoding片段的两大核心指标[3-4]。为了同时兼顾上述两方面需求,主流的eDNA metabarcoding片段(AcMDB07、Ac12S、MiFish-U、Tele02等)主要位于线粒体核糖体(12S、16S)高变区[3],虽然能基本满足鱼类多样性调查需求,但却存在着少数近缘鱼类因条形码片段变异速率较低而难以识别的风险。基于已有物种监测结果可知,采用核糖体区片段进行鱼类eDNA调查可能会造成“物种丢失”。如MiFish-U对平鲉属(Sebastes)、东方鲀属(Takifugu)、蝴蝶鱼属(Chaetodon)[7],Tele01对玉筋鱼属(Ammodytes)、杜父鱼属(Cottus)、雅罗鱼属(Leuciscus)[8]等类群的OTUs注释结果均出现了此问题。表明“物种丢失”可能是一种比较常见的现象。然而,由于本底资料缺乏、物种数量不足等原因,已知的鱼类eDNA调查只是简单提及了此现象,未能充分揭露所用片段的近缘物种识别缺陷;不同片段间的近缘物种识别差异更缺乏相互比较。究竟哪些片段的“物种丢失”风险更低,更适合近缘鱼类多样性调查?目前尚不得而知。
随着水体污染、过度捕捞、栖息地丧失等威胁的加剧,世界各地土著鱼类的物种多样性、遗传多样性正急剧下降[9-11];而外来鱼种却因水产养殖、观赏活动释放等原因大幅增加[11]。这就对当前的鱼类多样性监测提出了更高的要求。而全面、准确的物种调查则是开展各类生物多样性保护的基础和前提[1-3]。在近缘鱼类识别是鱼类eDNA调查无法回避的大背景下,本研究将系统探究主流metabarcoding片段的“物种丢失”风险、比较不同片段的近缘物种识别差异,以期能够从中筛选出物种识别率最高的片段,最终为完善鱼类eDNA metabarcoding技术、提高鱼类多样性调查结果的准确性提供一定技术支撑。
2 材料与方法
2.1 序列下载及筛选
自然水域难以出现近缘鱼类大规模出现的情况,结果具有较大的偶然性。因此,本研究采用MitoFish公共数据库内的线粒体全序比较各片段的近缘物种识别差异。具体操作如下:登录MitoFish数据库(http://mitofish.aori.u-tokyo.ac.jp/download.html),下 载“All Mitogenomes”选项内的3 081种鱼类的线粒体全序fasta压缩文件(截至2022年1月23日);同步下载txt文档,以方便核对序列来源、GenBank号、上传者单位等信息。使用MegAlign软件将下载的fasta文件转换为seq文件。基于Fishbase数据库(https://www.fishbase.de/)核对物种分类有效性。剔除杂交种、争议种及含有简并碱基的序列。为降低序列截齐的工作量,本研究仅将物种较多(5种及以上)的属挑出用于后续分析。物种分类阶元的确认依据Fishbase数据库及中国动物主题数据库(China Animal Scientific Database,http://www.zoology.csdb.cn/)。
2.2 metabarcoding片段选择
查阅2008−2021年期间涉及鱼类eDNA metabarcoding技术的有关研究,特别是参考Zhang等[3]的研究结果对其中表现较好的metabarcoding片段及对应引物进行汇总。以花鳗鲡(Anguilla marmorata)线粒体全序(序列号:NC006540)为参照,核对正、反向引物方向标注是否正确。按所在位置对metabarcoding片段进行编号、排序。
2.3 序列截齐
使用DNAstar软件包中的Seqman程序,对挑选出的序列进行截齐:(1)扩增片段序列截齐后用于近缘鱼类条形码相似度分析;(2)正、反向引物序列截齐后用于引物变异度分析。
2.4 遗传距离分析
采用PAUP 4.0软件构建最大简约法(Maximum Parsimony,MP)系统发育树,系统树可信度均采用Bootstrap检验,经1 000次重复抽样检验得到分支树节点支持率。物种的聚类情况即物种识别率[12]:若不同物种聚为一支(取遗传距离等于0),认为该物种识别失败;若不同物种聚为不同支(遗传距离大于0),认为该物种识别成功(由于不同metabarcoding片段的种间差异标准不同,难以给出准确可靠的判定阈值,因此本研究只探究遗传距离是否为0的情况);同时,基于正、反向引物联合序列计算总平均遗传距离[7],从而判断引物的通用程度。
运用SPSS25.0 软件基于单因素方差分析(Oneway ANOVA)对不同引物的物种识别率等进行差异显著性分析。显著差异阈值为p=0.05,试验结果以“平均值±标准差”表示。
2.5 非度量多维尺度分析
使用非度量多维尺度(Non-Metric Multidimensional Scaling,NMDS)[13]方法对物种识别结果 [0,1]矩阵进行排序分析,该分析在Canoco 5.0软件中完成。选用Bray-Curtis距离,因为其在大范围和小范围的坐标轴上都具有稳健性[12,14]。分析结果以胁强系数(stress)作为评判标准:当stress<0.2时,认为可以用NMDS的二维点图表示,该图形有一定的解释意义;当stress<0.1时,认为该排序是一个好的排序;而当stress<0.05时,则认为该排序结果具有很好的代表性[15]。NMDS运算的步骤如下:(1)将物种识别率处理为[0,1]矩阵,鉴定成功为1,鉴定失败为0;以物种名为首行、metabarcoding片段名为首列储存.xlsx格式的[0,1]矩阵;(2)打开Canoco 5.0软件,加载.xlsx文档,给定table name,选择“import all species as factors”,其余选择默认参数,完成矩阵导入;(3)“analysis”选项下,选择“Canoco Adviser”,调出 NMDS 分析程序;(4)“treatment of ties in distance”选择“secondary”, “stress formula”选择“type 1”,其余选择默认参数。
3 结果
3.1 用于分析的近缘鱼类简介
本研究用于分析的鱼类共计2纲、20目、52科、106属、935种。其中鲤形目(Cypriniformes)种类最多,共计3科、37属、337种,鲈形目(Perciformes)次之,共计 18科、22属、190种。扁鲨目(Squatiniformes)、脂鲤目(Characiformes)、胡瓜鱼目(Osmeriformes)的数量最少,各只有1科、1属、5种。物种名称及对应Genbank序列号见表S1、表S2。
3.2 引物简介
经文献查阅及汇总,特别是参照Zhang等[3]对22个主流片段(引物)的全面比较结果,本研究用于分析的鱼类eDNA metabarcoding片段共计15个。涵盖线粒体12S、16S、COI 3个目标基因。其中以12S为目标基因的片段最多(8个),16S次之(6个),COI最少(仅1个)。各片段、对应引物位置及信息见图1及表1。
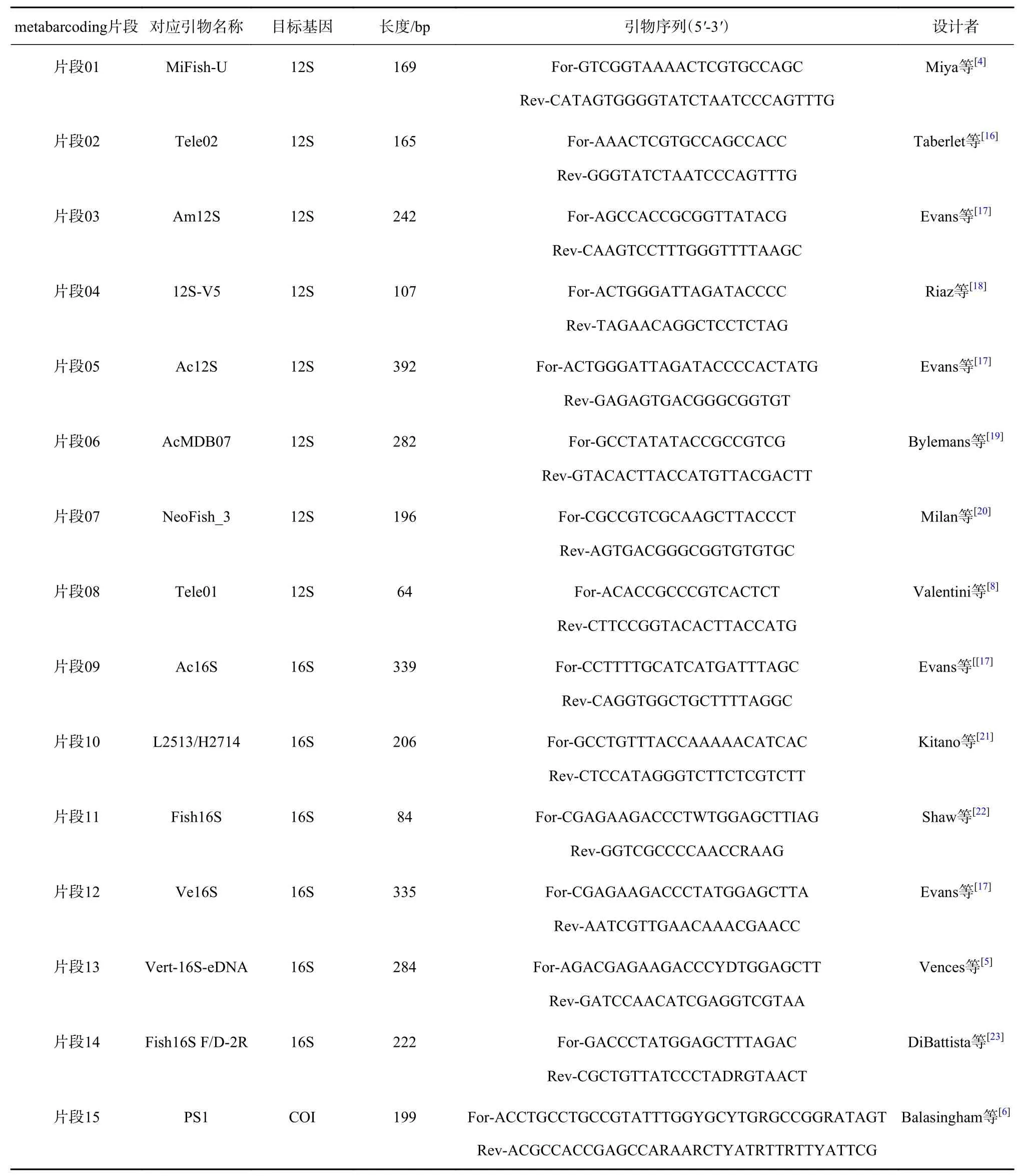
表1 本研究中15个鱼类eDNA metabarcoding片段简介Table 1 Summary of 15 fish eDNA metabarcoding fragments analyzed in this study
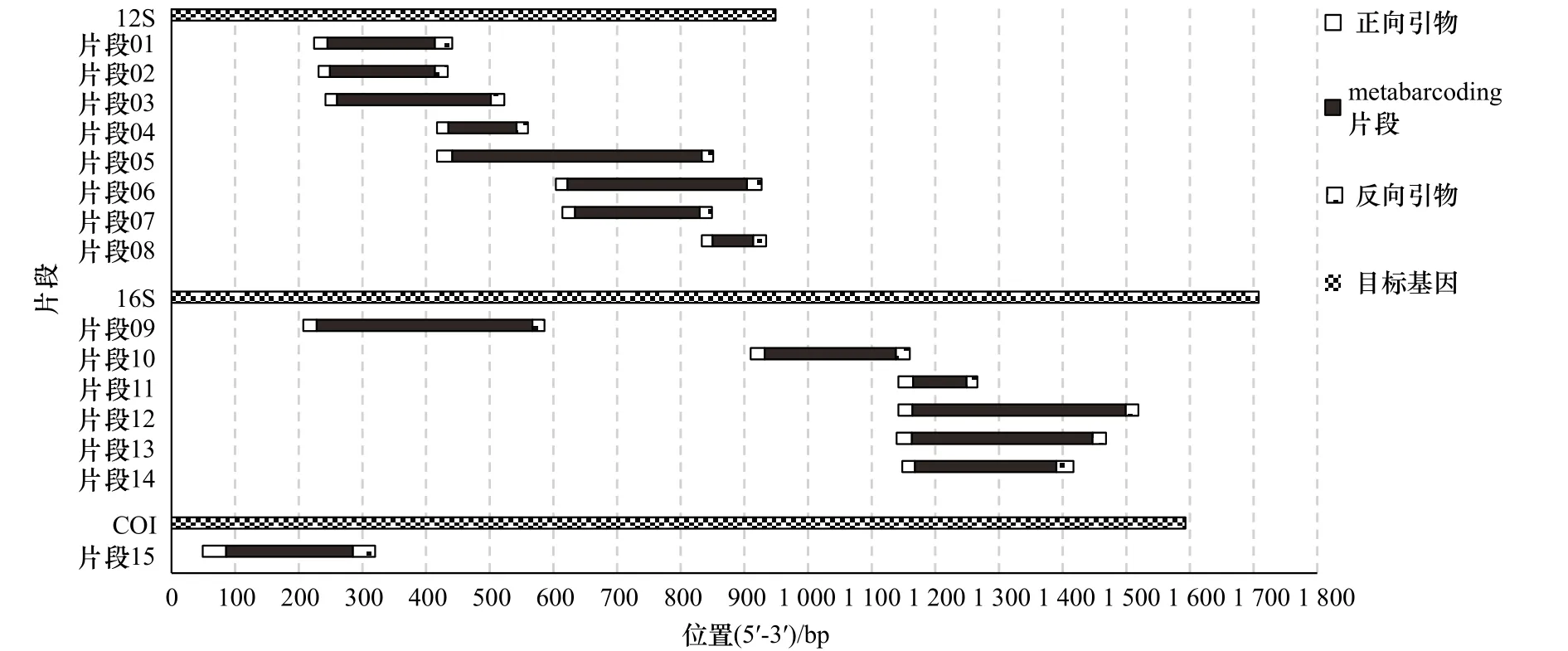
图1 本研究中15个鱼类eDNA metabarcoding片段和对应引物在目标基因上的位置Fig.1 Locations of the 15 fish eDNA metabarcoding fragments and primer pairs on the target mitochondrial genes
3.3 物种识别率及引物变异情况
不同片段的物种识别率存在极显著差异(F=32.39,p=1.5×10−76),蛋白质编码基因(片段 15)的物种识别率最高,达到(82.76±24.66)%。核糖体基因中,片段03、片段05的表现最为优异,识别率分别为(68.9±30.81)%和(71.62±27.3)%;片段 04、片段 08和片段11的识别率较低,平均值未超过42.92%;其余片段的物种识别率较为中等,平均值在54.58%~64.6%间(图2,表2)。
基于15个片段对应正、反向引物联合序列构建的MP系统发育树总平均遗传距离依次为1.52%、1.84%、4.58%、0.88%、0.97%、2.46%、5.78%、3.32%、9.32%、2.29%、6.21%、4.14%、1.71%、3.17%和15.70%(图2)。表明蛋白质编码基因(片段15)的对应引物通用性最差。片段09、片段11、片段07、片段03和片段12的引物变异程度也较高(总平均遗传距离≥4.14%),实际使用过程中应考虑其eDNA扩增效率。
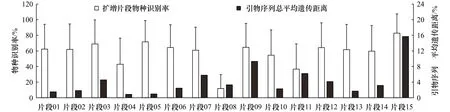
图2 本研究中15个鱼类eDNA metabarcoding片段的物种识别率及对应引物序列总平均遗传距离Fig.2 Fish species resolution rates of 15 eDNA metabarcoding fragments and overall mean distances of primer pairs in this study
不同类群的识别率也存在极显著差异(F=14.40,p=7×10−157):旗鳉属(Aphyosemion)、舌鳎属(Cynoglossus)、镖鲈属(Etheostoma)等属内近缘物种较容易区分,全部15个片段的平均识别率均在90%以上;而金枪鱼属(Thunnus) 、白鲑属(Coregonus)等类群的目标基因同源性较高,大部分片段对其识别能力较差(图3,表2)。
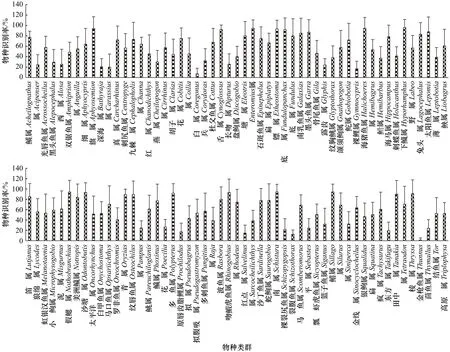
图3 本研究中106属鱼类的物种识别率Fig.3 Fish species resolution rates of 106 genera in this study

表2 metabarcoding片段对各类群的识别率Table 2 The species resolution rates of metabarcoding fragments for different groups
3.4 NMDS结果
根据935种鱼类的识别结果对15个metabarcoding片段做NMDS排序,片段04、片段08和片段11明显远离其余片段,说明此3个片段物种识别结果较为不同(图4)。其余11个核糖体片段根据所在目标基因聚类成A、B两组,表明不同目标基因影响物种识别。NMDS分析结果的胁强系数为0.13,说明将15个metabarcoding片段划归不同组具有一定的解释意义。
4 讨论
本研究用于分析的鱼类数量庞大、阶元众多(见3.1节及表S1、表S2),涉及软骨鱼类及硬骨鱼类、淡水鱼类及海水鱼类、定居性鱼类及洄游性鱼类等各类群。样本量具有较好的统计学意义,结果能够比较充分地反映15个主流eDNA metabarcoding片段的优劣。本研究是目前国内第一篇针对性探究鱼类eDNA metabarcoding近缘物种适用性的报告。
4.1 COI片段的引物缺陷
片段15(PS1,COI)的物种识别率显著高于剩余14个12S/16S片段,是唯一物种识别率超过80%的metabarcoding片段。这与Balasingham等[6]认为的鱼类线粒体蛋白质编码基因比核糖体基因能够更有效避免北美五大湖内部分入侵物种eDNA注释结果假阴性的结论一致,也与经典的DNA条形码普遍采用线粒体细胞色素C氧化酶亚基I基因的事实相符[24]。此外,COI基因在鱼类eDNA metabarcoding参考数据库的全面性方面也具有12S、16S基因难以比拟的优势[3]。然而,引物序列的高变性可能会阻碍片段15的广泛应用。本研究中该片段正、反向引物序列的总平均遗传距离高达15.70%。中国团扇鳐(Platyrhina sinen-sis)、细条银口天竺鲷(Jaydia lineata)、土佐鰧(Uranoscopus tosae)、横带髭鲷(Hapalogenys analis)的 COI特异性引物、探针设计结果表明:当总平均遗传距离大于8.21%~9.65%时,高纯度DNA模板会出现明显的弥散扩增;总平均遗传距离大于12.47%~13.77%时,引物会对部分近缘物种产生绝对特异[7]。因此,片段15存在eDNA低效扩增/偏倚扩增的缺陷。Collins等[25]、Zhang等[3]分别对英吉利海、北京水系的鱼类多样性调查显示,COI片段的物种检出数目不及所用的12S片段,其结果支持本研究观点。
此外,其他的COI或Cytb metabarcoding片段可能也难以解决引物通用性的问题。鱼类eDNA metabarcoding技术需要依托 2×150 bp、2×250 bp两种高通量测序(Next-generation Sequencing,NGS)平台实现[3],而Miya等[4]对880种硬骨鱼类线粒体基因组全序的筛查结果显示,COI、Cytb等蛋白质编码基因在该读长限定范围内不存在成对的侧翼保守区,理论上难以设计出与经典的鱼类DNA条形码(基于Sanger测序,读长可达2×600 bp)媲美的成对引物。Menning等[26]、Jennings等[27]、Sultana等[28]的序列比对结果也表明,整个Cytb基因基本为高变序列,COI基因则只在近5′端前350 bp范围内变异较小,相对适合设计短片段通用引物。结合本研究(图1)和Zhang等[3]对22对引物的汇总结果可知,片段15等少数几对鱼类COI metabarcoding优质片段恰位于此相对保守区,其引物通用性较差,而其他区域metabarcoding片段的引物序列总平均遗传距离可能更大。因此,本研究认为后续研究没有必要进一步比较其他COI、Cytb片段的近缘物种识别差异;同时认为以片段15为代表的蛋白质编码片段更适合作为eDNA研究的辅助metabarcoding,并且需要根据研究水域、目标类群降低引物调查范围,从而对引物序列进行针对性设计或优化。
4.2 核糖体片段的比较
片段长度影响物种识别(表1,图2,图4)。本研究中识别率最低的3个metabarcoding片段(片段08、片段11和片段04)长度均不超过110 bp,而片段05、片段03等高识别率片段则在200 bp以上。Balasingham等[6]、Gantner等[29]的研究也认为12S和16S扩增片段越短,物种鉴定的准确性越低。虽然由于水体中的痕量eDNA通常存在严重降解,主流的观点认为200 bp以内的微条形码可能具有更高的PCR成功率[4,30],但Bylemans等[19]、Zhang等[3]多个研究却发现:片段05、片段03等的高通量测序数据量和物种检出数目并不低于片段08、片段04或片段11,只是在定量分析等方面相互存在较大差异。鱼类eDNA的产生、降解动力学过程极为复杂,目前没有研究能够准确阐明eDNA的微观存在、分布和变化规律,Deiner等[31]、Bylemans等[19]推测长片段eDNA可能反而可以更长久地存在水体中。本研究不推荐使用metabarcoding短片段(片段08、片段11和片段04)进行近缘鱼类多样性调查。
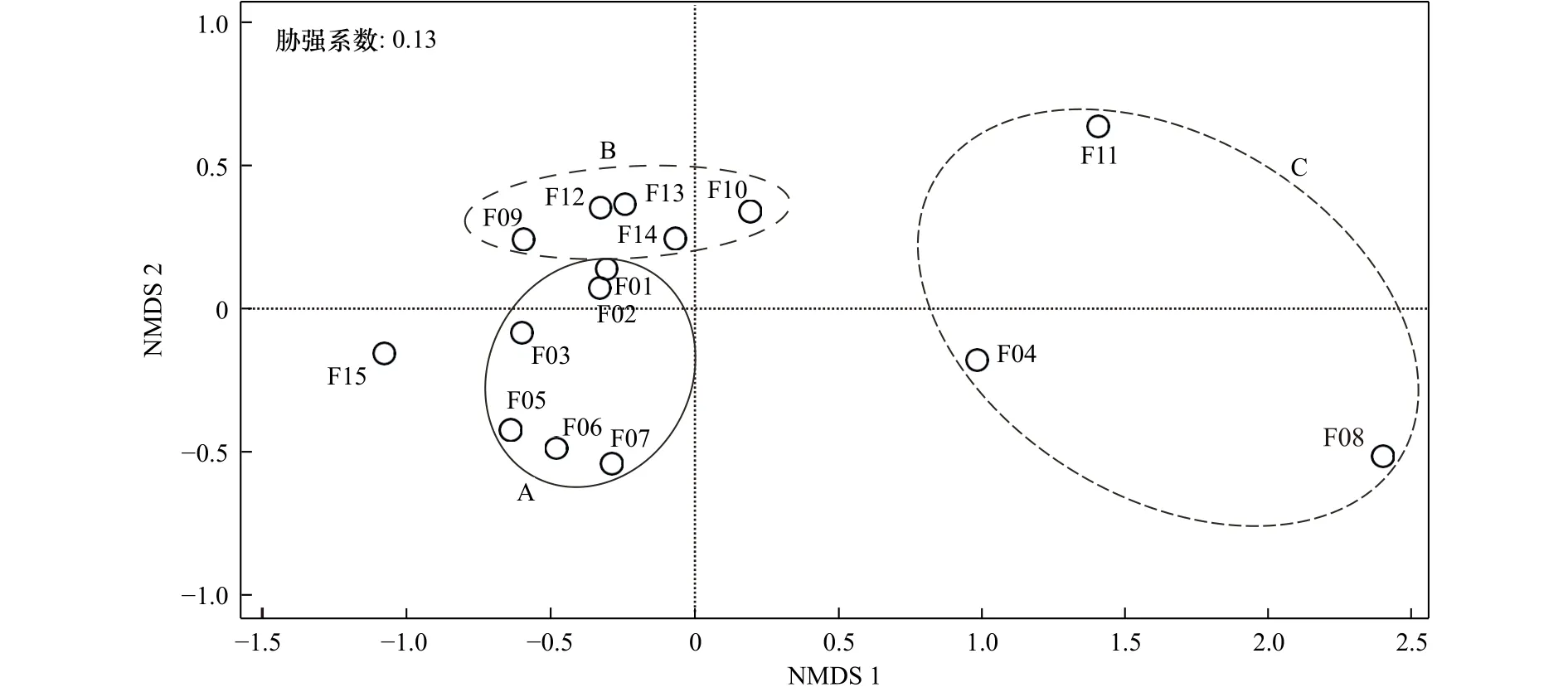
图4 不同metabarcoding片段的非度量多维尺度(NMDS)分析Fig.4 Analysis of non-metric multidimensional scaling (NMDS) for different metabarcoding fragments

续表2
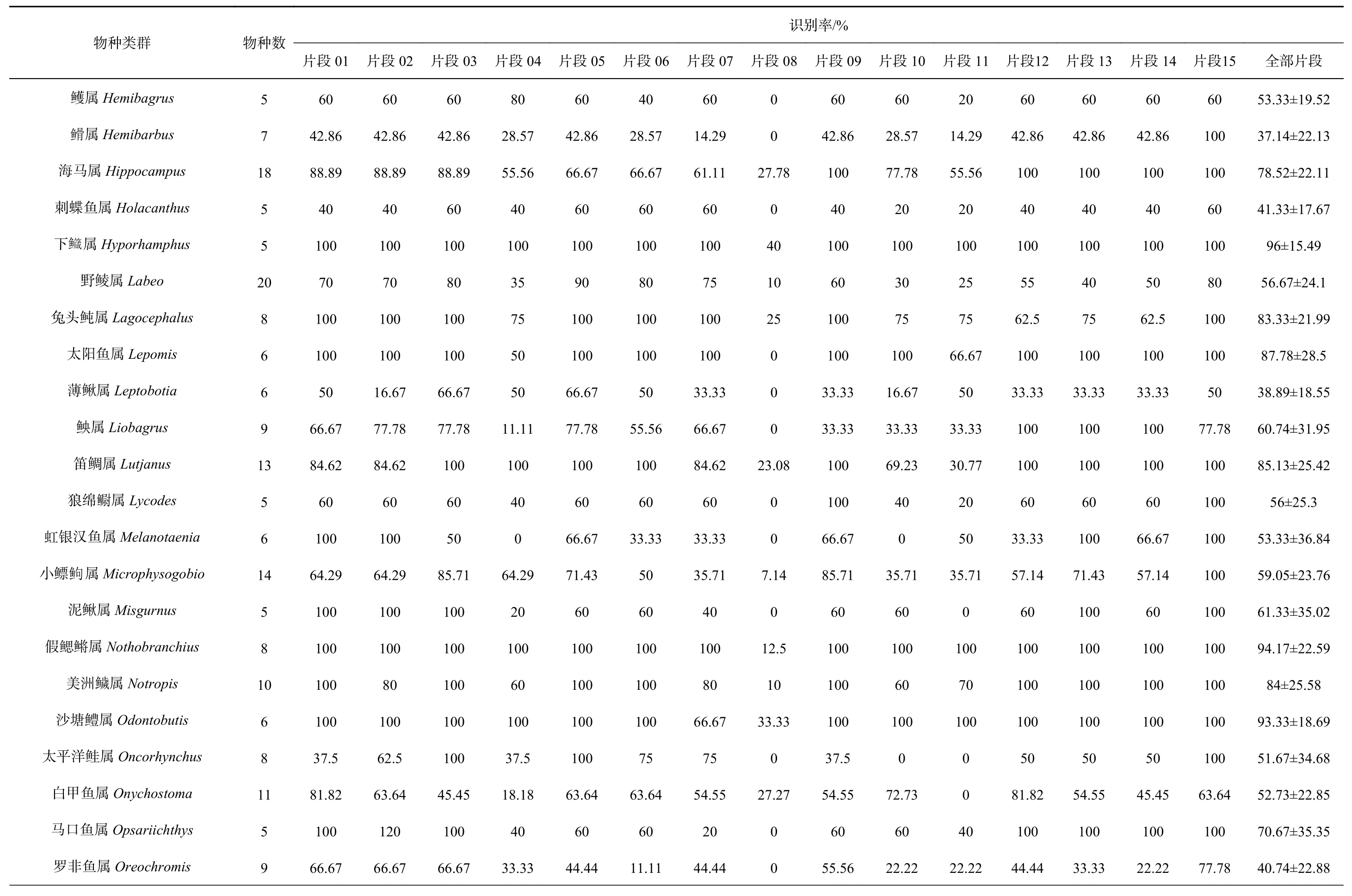
续表2
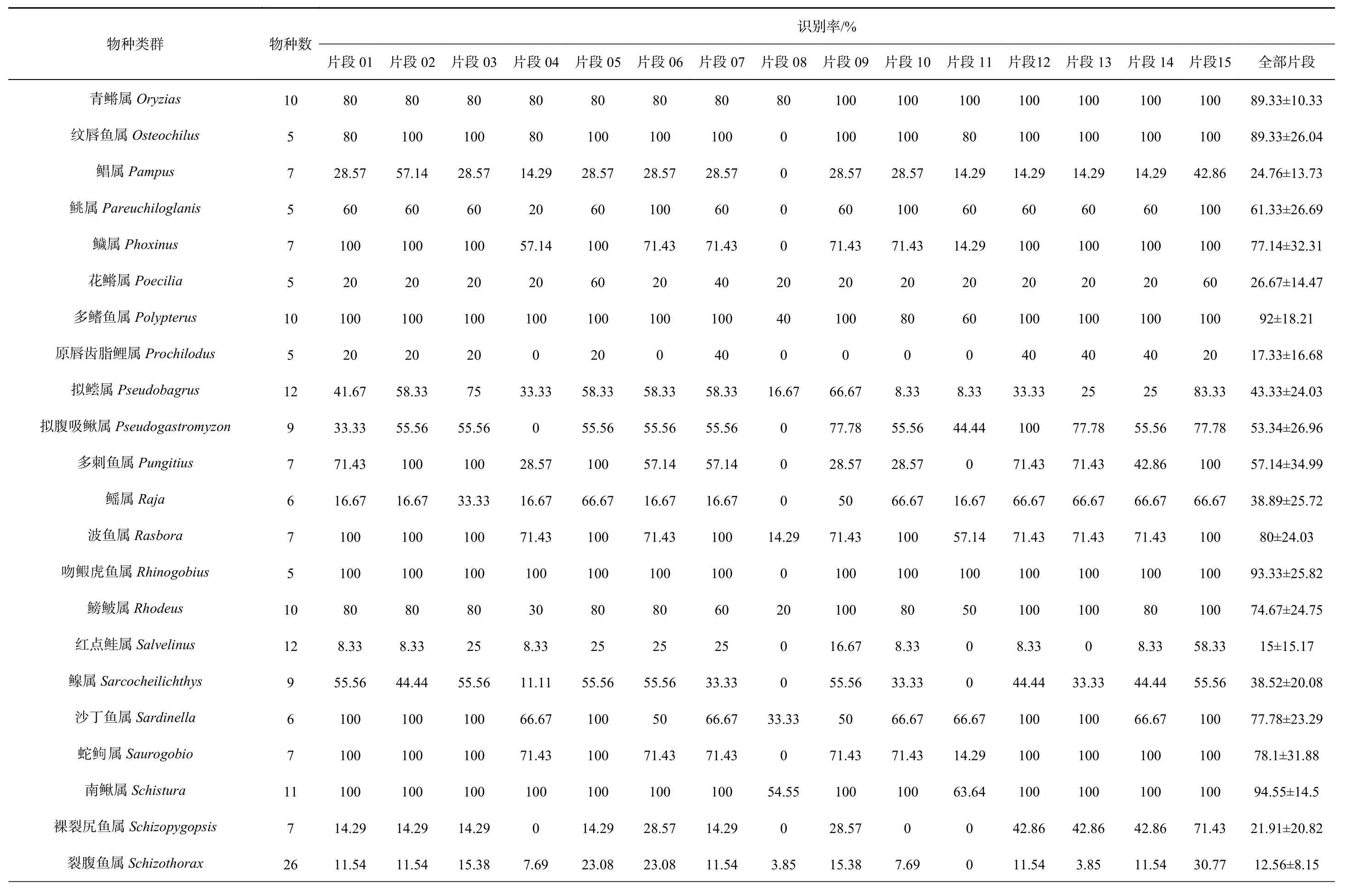
续表2
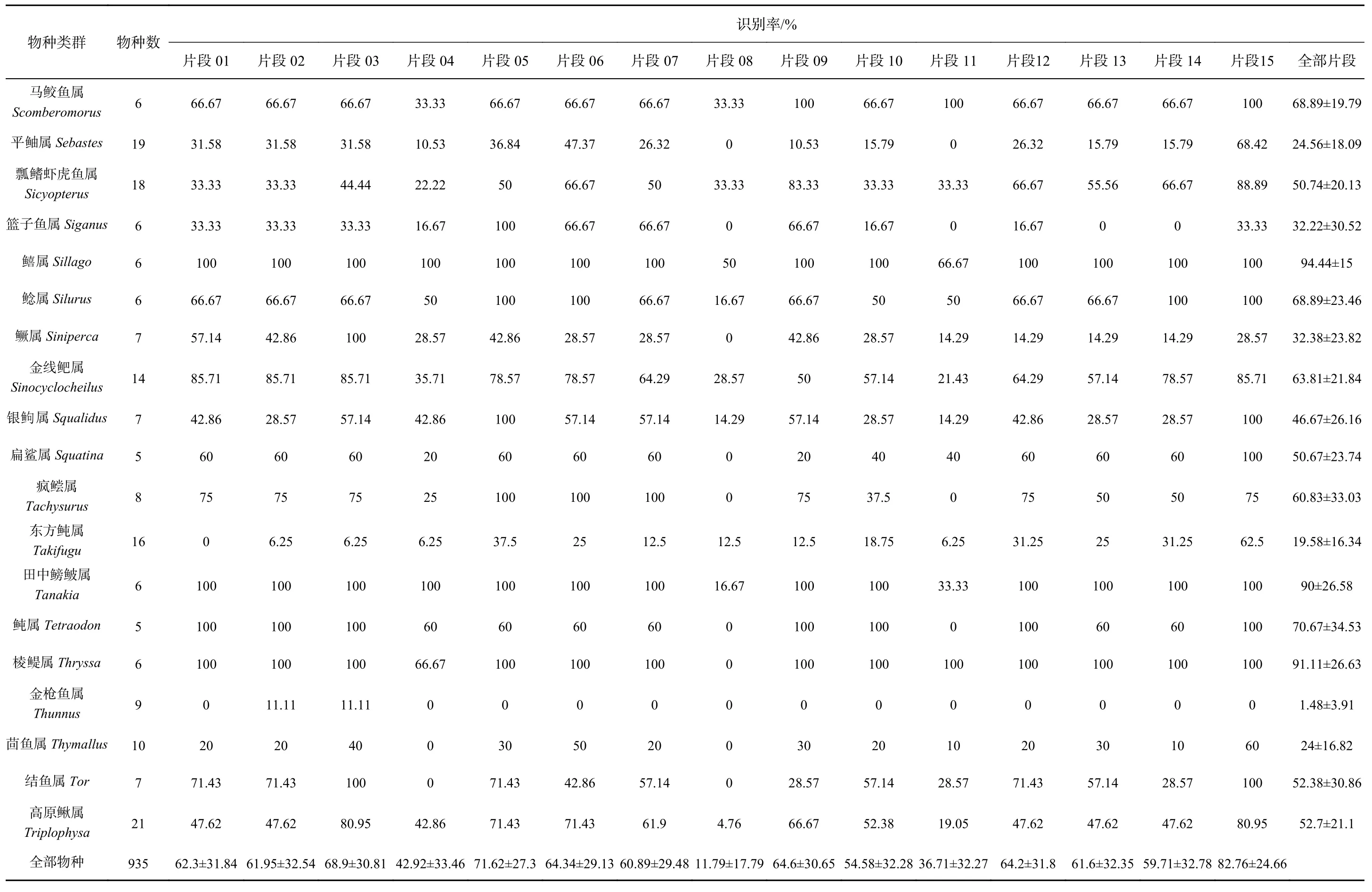
续表2
除去COI和3个短片段,片段05的物种识别率最高,其引物通用性(总遗传距离=0.97%)也仅次于片段04。片段05是近缘鱼类多样性调查的第一选择,该结果与Zhang等[3]的研究结论一致。片段09、片段07、片段03、片段12的物种识别率虽然也较高,但在本研究中,其引物序列总平均遗传距离较大(≥4.14%),存在类似于片段15的“物种丢失”风险;其他metabarcoding引物比较研究也表明其物种检出数目不及片段06、片段13[3]。片段14、片段10虽然长度大于200 bp,但物种识别率和引物通用性却并不出众;并且可能因为引物序列3′位置存在变异[7],这两个片段在个别研究中的实际表现甚至不如片段15[3]。而片段01和片段02则在引物序列、条形码性能方面基本一致,二者存在明显的相互替代性,本研究更倾向二者中选用片段01。综上所述,片段05、片段06、片段01、片段13是本研究筛选出的4个最优metabarcoding片段。然而,受高通量测序成本和参考数据库的影响,在单一研究中使用的metabarcoding片段通常不超过3个[3-4]。虽然本研究及Zhang等[3]、Bylemans等[19]的研究均表明片段06的物种识别率和引物通用性优异,但NMDS分析(图4)显示片段06在散点图上与片段05位置较近,因此同时使用这两个片段进行鱼类多样性研究可能并不能显著提高物种检出数目。NMDS分析也显示,不同基因、同一基因不同片段间均存在较大差异(图4)。因此不仅需要多片段联合应用,而且所用片段要有足够的多基因代表性。本研究倾向于以片段05、片段01为主,片段13等为辅,进行近缘鱼类多样性调查。
4.3 本研究所用片段的不足及新片段开发的可能性
本研究对106属鱼类的识别结果差异极显著(见3.3节),表明物种类群直接影响eDNA metabarcoding调查效果。从15个片段的总识别率角度比较(表2),仅15属鱼类物种识别率超过90%,多达38属鱼类识别率不足50%。由于935种鱼类的线粒体全序列皆来自公共数据库,其中一些序列可能存在同物异名错误—如鲳属鱼类的银鲳(Pampus argenteus)与镰鲳(Pampus echinogaster)[32],因此该属鱼类识别率偏低;但更可能的解释是低识别类群物种间遗传差异较小,导致metabarcoding片段从源头上就难以完全区分—如白鲑属(Coregonus)[33]、红点鲑属(Salvelinus)[34]的众多物种集中分布于50°N以北环北极圈水域,物种间可能存在较为频繁的基因交流;平鲉属(Sebastes)则广泛分布于北太平洋且仔鱼营漂浮生活,不同群体间也不存在明显的地理隔离[35]。Miya等[4]在筛选Mi-Fish-U(片段01)引物时已发现,金枪鱼属等大洋性鱼类的大部分线粒体基因高度保守,只有借助变异速率极高的线粒体NADH脱氢酶亚基5(NADH Dehydrogenase Subunit 5,ND5)条形码才能进行有效区分。因此,受制于物种自身的遗传背景,本研究所用片段尚无法对全部鱼类进行100%区分。后续仍需不断尝试筛选新片段从而尽量提高eDNA metabarcoding技术的鱼类多样性调查能力。
metabarcoding片段的开发与目标基因内茎环结构的数量和分布情况密切相关。斑鰶(Konosirus punctatus)[36]、大口鰜(Psettodes erumei)[37]、北极茴鱼(Thymallus arcticus)[38]等线粒体基因二级结构示意图显示,16S基因仅在中后段存在1~2个大型茎环结构,相比之下,12S基因存在2~4个。由于12S基因的总长度仅为16S的60%(约900 bp : 1 600 bp),因此该基因内的茎环结构分布更为密集和均匀。茎环结构有利于寻找稳定的“保守−高变−保守”序列,Evans和Lamberti[39]、Hänfling 等[40]、Bylemans等[19]据此认为,线粒体12S基因比16S基因更适合开发鱼类eDNA metabarcoding标记,本研究支持此结论。然而,基于14个核糖体片段的位置示意图(图1)和简约信息(表1)可知,鱼类16S基因的后半段大型茎环及12S基因的主要茎环皆已经被筛选出metabarcoding片段。围绕单一茎环结构开发新片段不仅可能性极小,而且片段识别率可能也不理想(如片段07、片段10)。相比片段01、片段04和片段08等,片段05和片段06等则是由多个中小型茎环结构组合形成,其物种识别率反而更高。以此类推,联合多个相邻茎环区域可能是形成新的高识别率片段的手段之一。本研究中,12S片段对应引物存在明显的位置重合及序列共用(图1,表1),说明这些区域的引物保守性得到了较大认可。因此,可以尝试通过这些引物的组合应用开发更长、更高识别率的metabarcoding片段。如片段01、片段02的正向引物与片段06、片段07正向引物的反向互补序列共用,PCR产物约420 bp(图1)。该片段由1个大型茎环加2个小型茎环结构组成,其物种识别率可能高于片段05。
综合物种识别率、引物通用性等多方面因素,本研究推荐2×150 bp测序平台使用片段01(MiFish-U)、2×250 bp 测序平台使用片段 05(Ac12S),辅以片段13(Vert-16S-eDNA)进行近缘鱼类多样性调查;新片段的开发也是不断完善eDNA metabarcoding技术的重要工作。然而,鱼类eDNA研究受多方面因素的影响,基于935种近缘鱼类线粒体全序的比较结果可能会与实际表现有差异。后期还需寻找近缘鱼类广泛分布的水域,在建立庞大的本底资料数据库的基础上验证本研究的结论和推测。
补充材料
表S1 本研究中106属鱼类简介
表S2 本研究中935种鱼类学名及对应序列号
补充材料可通过https://www.hyxbocean.cn/获取。补充材料未进行排版和编辑,内容的准确性和科学性由作者承担。
