无需测序的两步法鉴定遗传变异肽的工具开发
2022-08-16宋丽丽宋娜娜邢清和
李 欣 宋丽丽 宋娜娜 邢清和 周 峰,3△
(1复旦大学生物医学研究院 上海 200032;2复旦大学附属妇产科医院新生儿科 上海 200090;3复旦大学附属中山医院肝癌研究所 上海 200032)
单 核 苷 酸 多 态 性(single nucleotide polymorphism,SNP)指基因组上由单个核苷酸的变异所引起的DNA 多态性,由SNP 引起的蛋白质中氨基酸序列发生变化的肽段称为遗传变异肽(genetically variant peptide,GVP),鉴定GVP 对于了解个体特异突变和潜在疾病等有重要作用[1-2]。蛋白质组学工作流程通常依赖参考数据库来识别肽段和蛋白质,如果数据库中不包含突变序列,就无法检测到GVP[1,3]。
得益于测序技术的快速发展,利用参考基因组或表达序列标签(expressed sequence tag,EST)的六框或三框翻译来构建包含所有GVP 的数据库成为可能,但是会使数据库大小急剧增加而导致结果中假阳性过高[1,4]。利用样品对应的测序数据生成定制的蛋白质序列数据库可以避免上述问题[5-7],但需要耗费额外的成本,且依赖于复杂的生物信息学分析。在某些场合我们甚至难以获取足量的核酸样品用以测序,比如犯罪现场的毛干[8]。
随着技术不断进步,大规模检测遗传多态性可能成为蛋白质组学数据分析流程中的常规工作。针对GVP 检测,目前尚缺乏不依赖对应样品高通量测序的有效工具[1-2,9-10]。Pratik 等[11]证明在蛋白质组学数据分析中运用两步法的迭代搜索策略可以有效提高肽谱匹配的灵敏度,改善搜索结果。本研究在此基础上开发了基于两步法的迭代搜索策略来鉴定GVP 的工具2Steps_GVPtool,并在不依赖对应样品高通量测序数据的基础上,以公共数据库中的遗传变异信息和参考蛋白质数据库构建了包含GVP 的数据库。考虑到实际应用需求,我们以毛干样品为例进行了实验。
材料和方法
数据采集采集一名女性志愿者的长发,志愿者对研究内容知情同意,年龄18~45 岁,既往无脱发史,无头面部手术史,无放化疗史,近一年内头发未烫染且可提供的头发长度>5 cm。剪掉长发首尾两端各2 cm,保留中间毛干部分,分成共5 mg 的4 等份。每一份单独按照文献方法[10]进行碎裂、酶解,处理后得到肽混合物,以4 标iTRAQ 对4 份生物重复样品进行标记之后混合,样品进样和数据采集过程采用实验室开发的全自动全蛋白质组定量分 析 平 台[12],所 用 质 谱 仪 为SCIEX 公 司 的TripleTOF 5600。
输入文件2Steps_GVPtool 是一个命令行驱动的软件工具包,可以在Linux 环境下作为独立应用程序安装使用,工具包储存在GitHub(https://github.com/lx18211510001/2Steps_GVPtool),方便用户下载使用,也可以集成为系统工程流程的一部分。2Steps_GVPtool 的输入文件包括遗传变异信息和参考蛋白质数据库,本研究使用dbSNP 数据库中的遗传变异信息和SwissProt 数据库中的参考蛋白质数据库。美国国家生物技术信息中心为满足基因组变异通用目录的需求而建立了dbSNP 数据库,存储了多个物种的全部变异信息,我们以dbSNP 中人类目录下的common_all_20180418.vcf.gz(https://ftp.ncbi.nih.gov/snp/organisms/human_9606/VCF/)作为输入文件,该文件包含所有至少一个主要群体中最小等位基因频率(minor allele frequency,MAF)≥0.01 的变异。
数据分析数据库构建和GVP 鉴定均通过2Steps_GVPtool 进行,具体参考工具包中的操作步骤。搜库软件使用ProteinPilot V.4.5(AB Sciex),肽谱匹配(peptide spectrum match,PSM)的错误发现率(false discovery rate,FDR)设定为0.01。
基因组序列验证使用DNeasy®试剂盒(美国Qiagen 公司)从个体血样中分离基因组DNA,对于2Steps_GVPtool 鉴定到的变异肽,使用TransStart®TopTaq DNA Polymerase(全式金)扩增编码蛋白质序列的相应扩增子,进行PCR 反应,通过Sanger测序获得扩增子序列。
结 果
流程框架本工具包的工作流程(图1A)包括变异位点注释(a 部分)、蛋白质组学数据分析(b 部分)和GVP 鉴定(c 部分)。在a 部分中,使用变异注释软件对已有的变异位点信息进行注释,挑选出非同义SNP 并生成每个SNP 突变前后对应的两条蛋白质序列;在b 部分中,对蛋白质组学数据进行常规的搜库分析,得到该样品包含的蛋白质及相对丰度信息;在c 部分中,利用前两步得到的信息,在常规的参考数据库中加入样品中高丰度蛋白质的GVP序列,生成新的数据库并再次搜库分析,得到样品中的GVP 信息。b 部分是非必须的,缺少b 部分时,构建的数据库会包含所有蛋白质的GVP。基于SwissProt[15]中的人类参考蛋白质序列数据库和dbSNP[16]中的人类常见变异信息,我们构建了一个通用的数据库ComVarDB(common variation database),即常见的变异数据库。ComVarDB 中包含17 484个蛋白质的168 000 个独特GVP 和SwissProt 的20 365 个序列(图1B),GVP 序列使ComVarDB 相比于原始SwissProt 数据库增加了约19.2%。

图1 工具包工作流程和数据库比较Fig 1 Workflow of toolkit and composition of the three databases
GVP 鉴定我们以采集的毛干蛋白质组学数据对工具包2Steps_GVPtool 进行测试,4 份生物重复样品单独进行样品制备,iTRAQ 114/115/116/117 标记之后混合(图2A)。以SwissProt 的参考数据库搜库,共鉴定到2 350 个蛋白质(图2B)和6 935个独特的肽段,鉴定到的蛋白质数量远多于之前毛干蛋白质组学鉴定到的182~578 个蛋白质[2,10]。蛋白质数量大大提升得益于本课题组开发的全自动全蛋白质组定量分析平台,该系统是蛋白质组学第一次在蛋白质检测水平上达到与二代基因组测序技术同样的测定深度[12-14]。以ComVarDB 搜库共鉴定出16 个GVP,其中14 个得到Sanger 验证(表1)。鉴定出的GVP 大部分(11/14)都来自角蛋白和角蛋白相关蛋白,这与Chu 等[17]的研究一致,证明毛干中GVP 主要来源于角蛋白和角蛋白相关蛋白。大部分氨基酸之间的转变没有出现,在通过验证的14 个GVP 中,丙氨酸到脯氨酸的突变出现了2 次,丝氨酸相对更容易检测到突变(3/14),谷氨酰胺、亮氨酸、蛋氨酸、脯氨酸和丝氨酸均检测到2 次突变(2/14)。
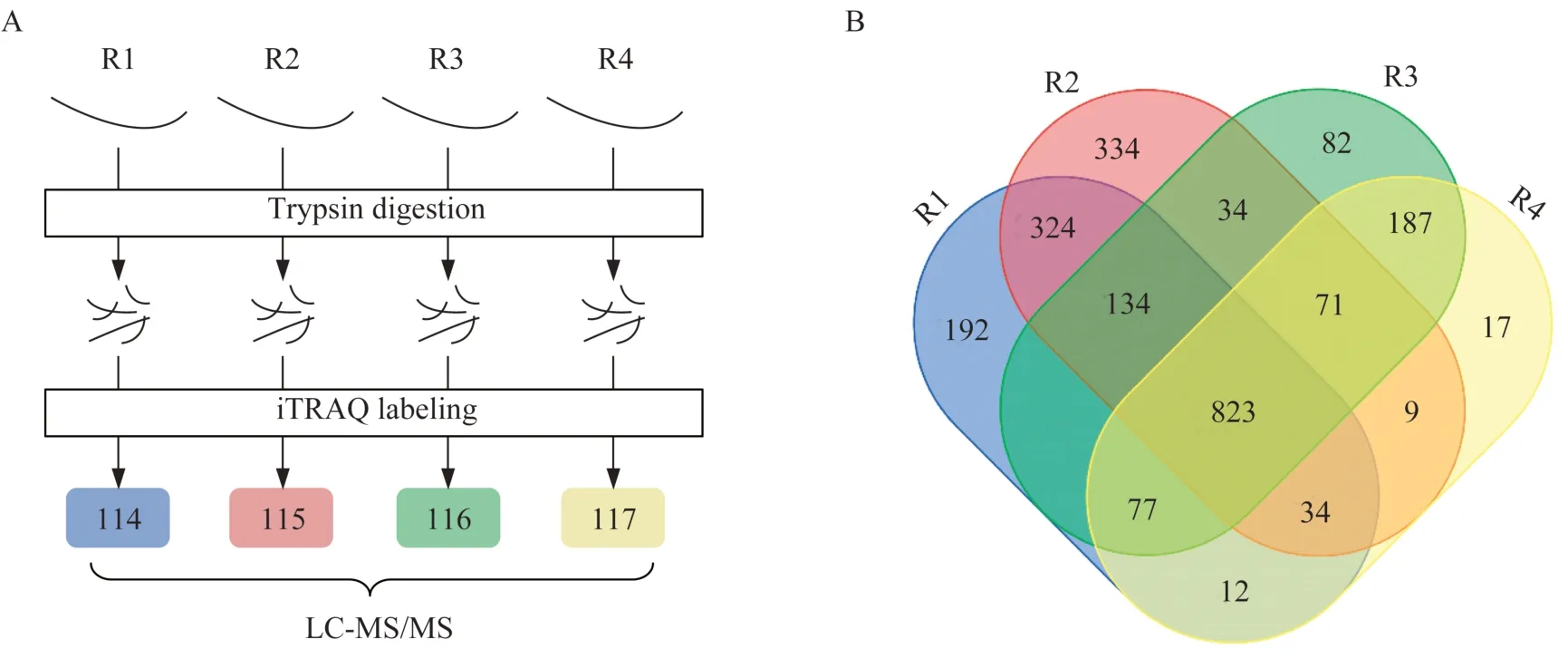
图2 实验流程和蛋白质韦恩图Fig 2 Experimental workflow and Venn diagram of protein identified

表1 ComVarDB 搜库鉴定到的GVPTab 1 GVPs identified by search against ComVarDB
数据库优化在蛋白质组学的实验中,表达量更高的蛋白质序列覆盖度更高,因而更有机会检测到高表达量的蛋白质中存在的GVP。因此,我们考虑使用第一步的搜库结果来对数据库进行优化,即在参考数据库中仅加入一些高丰度蛋白质的GVP序列,以减少数据库大小,进一步降低搜索空间。我们创建了7 个缩小的、包含GVP 的数据库:topAllVardb、top2000Vardb、top500Vardb、top200 Vardb、 top50Vardb、 top25Vardb、 top10Vardb。topAllVardb 表示数据库包含第一步搜库结果中检测到的全部2 350 个蛋白质的GVP,top2000Vardb表示数据库中包含第一步蛋白质鉴定中表达量排名前2 000 的蛋白质的GVP,以此类推,top10Vardb表示数据库中包含第一步蛋白质鉴定中表达量排名前10 的蛋白质的GVP。结果表明,各个数据库鉴定到的 GVP 重叠度很高,ComVarDB、topAllVardb 和top2000Vardb 成功检测到相同的14个GVP(图3A),检测到的GVP 数量在top500 Vardb 中达到最大值18(图3B),同时top500Vardb检测到的GVP 的准确率(验证正确的GVP 数量/检测到的全部GVP 数量)也是除top10Vardb 之外最高的(图3C)。
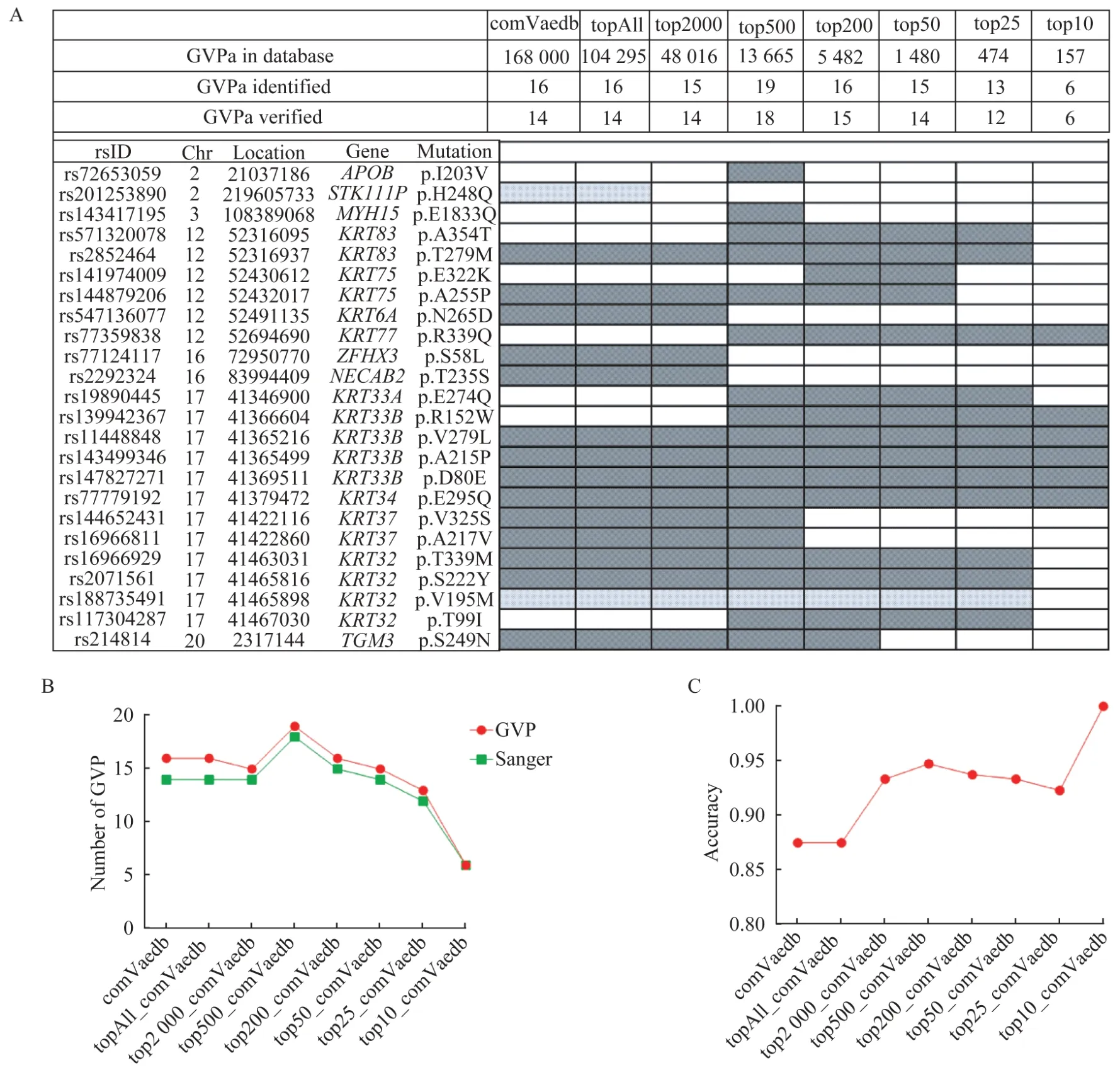
图3 数据库优化结果Fig 3 Results of database optimization
讨 论
在包含了大量氨基酸突变序列的蛋白质组数据库中,肽段被错误鉴定为遗传变异肽的风险很高[9,18]。本研究仅选择高频(MAF≥0.01)突变,严格控制了构建的数据库大小,以达到降低搜索空间的目的,在数据库中加入全部蛋白质突变时构建的ComVarDB 数据库,相较于原始参考数据库的增加幅度为19.2%,远低于其他通过公共数据库信息构建的参考蛋白质数据库至少翻倍的增加幅度[4,19]。数据库大小和搜索空间得到控制的同时,我们使用严 格 的FDR 阈 值(0.01)来 控 制PSM 质 量,ComVarDB 数据库的鉴定结果中GVP 确认率为14/16,高于此前GVP 研究中FDR 为0.05 所对应的确认率(6/9)[9]。我们同样检查了16 个GVP 对应的谱图,发现大多数谱图和肽序列的理论谱图匹配良好,而假阳性GVP 的谱图匹配则相差很多。手动检查GVP 谱图可能是GVP 验证的有效方案,已开发的工具如SpectrumAI[20]等可以实现谱图检查。
在蛋白质基因组学研究中,Muth 等[21]证明使用大型数据库进行初步搜索后,基于此搜索结果中PSM 对应的蛋白质来构建一个较小的数据库可以提高对样品中蛋白质的识别率。因而我们可以利用两步法的迭代的搜索流程来提高检测到的GVP数量和质量,我们不仅仅是构建了包含第一步搜索结果中全部蛋白质的GVP 序列的数据库,而是进一步利用仅在数据库中包含部分高表达蛋白质的GVP 序列,以达到再次降低搜索空间的目的。结果证明减少数据库中GVP 的数量的确有助于识别到样品中更多的GVP,其原因可能在于搜索空间的大小对谱图匹配的得分有很大影响。蛋白质组学数据库越大,库中的相似序列越多,当谱图匹配到这些相似的序列时,搜库软件会给出更高的罚分。每个数据集的数据质量和大小不同,对数据库中应该包含多少蛋白质的变异肽难以得出统一的结论,研究人员可以根据搜库结果进行调整以获得最佳选择。对于有多次样品需要分析的情况,本研究采用的是以所有样本间平均表达量的顺序为准来挑选高表达蛋白质,出于鉴定更多GVP 的目的,也可以考虑使用多个样品中高表达量蛋白质的并集来建库搜索。
鉴定GVP 对于从蛋白质层面了解个体特异的和潜在的疾病突变、进行精准治疗和个体鉴定等都十分重要[1],本研究构建的2Steps_GVPtool 可以针对所有细胞、组织、物种构建定制化的蛋白质基因组学数据库,用以检测其蛋白质组学数据中的GVP。在2Steps_GVPtool 工作流程中引入的基于两步的迭代的数据库搜索方法,可以达到在同样的数据集中检测到尽可能多的GVP。与从表达序列标签和参考基因组的翻译相比,该工具包大大减小了所构建的数据库大小,降低了搜索空间[1,4];与其他依赖样品测序数据来构建定制化数据库的工具相比,该工具包不需要测序等额外的耗费[5-7],且使用方法简单,工具包轻便易于整合到其他系统分析流程中。值得注意的是,本研究是通过仅在数据库中加入高表达蛋白的GVP 序列来缩小搜索空间和数据库规模,鉴定出更多的GVP 结果,这意味着我们主动丢失了低丰度蛋白的GVP 信息。对于低丰度蛋白的GVP 鉴定,建议按照图1A 中的a、c 两个步骤使用本工具,即不进行低丰度蛋白质的过滤或使用鉴定到的全部蛋白质。
随着技术不断进步,大规模检测遗传多态性可能成为蛋白质组学数据分析工作中的常态,为从蛋白质组学数据中获得的信息提供新的维度。然而,直接从蛋白质组学数据中获取GVP 信息并非易事。2Steps_GVPtool 作为轻量级的脚本工具包,用户可以在Github 直接获得完整工具,为鉴定蛋白质组学数据中的变异信息而生成定制的蛋白质序列数据库。一方面,用户可以通过公共数据库(如dbSNP、1000 Genome 等)中已有的变异信息来生成数据库;另一方面,在有条件获取样品对应的NGS 测序数据时,该工具包同样可以根据对应的测序数据来生成数据库。另外,在2Steps_GVPtool 中3 个步骤紧密连接的同时,工具包中各脚本之间的依赖性不强,所需的输入文件和输出文件均为有固定格式的文本文件,方便用户将单个脚本集成到更大型或其他用途的数据分析流程中。随着新一代测序技术和蛋白质组学的不断进步,该工具包将发挥重要作用。
作者贡献声明李欣 数据统计和分析,制图,论文构思、撰写和修订。宋丽丽 数据采集,论文修订。宋娜娜 数据采集。邢清和 论文指导和修订。周峰 论文选题、指导和修订。
利益冲突声明所有作者均声明不存在利益冲突。
