水合肼制氢纳米催化剂改性制备及机理研究进展
2022-08-10刘晓涯王金超刘莹马敬环
刘晓涯,王金超,刘莹,3,4,马敬环
(1 天津工业大学环境科学与工程学院,天津 300387; 2 华电水务工程有限公司,北京 100071;3 精馏技术国家工程研究中心,天津 300072; 4 北洋国家精馏技术工程发展有限公司,天津 300072)
引 言
能源是人类生存和发展不可或缺的物质基础,其中氢能[1-3]因其能量密度高、清洁无污染、能量转化效率高、通用性强而成为最具发展潜力的新能源之一。质子交换膜燃料电池(proton exchange membrane fuel cells, PEMFCs)能够将氢的化学能直接转化为电能,不受卡诺循环效应的限制,具有发电效率高、无污染、无噪声、冷启动快以及比功率高等优点,加速了氢能的高速发展,但是安全高效、响应迅速的在线氢源是实现PEMFCs 商业化应用的先决条件和关键技术[4-5]。目前制氢的研究热点有电催化制氢、光催化制氢、光电催化制氢和新型化学储氢材料催化制氢等[6]。电催化分解水制氢是利用电能作为能量来源,通过外加电场的作用分解水生成氢气,具有产物纯度高、无污染、制氢效率高等优点。但由于电解过程中电极反应的过电位高导致的电能消耗大,以及电解装置的成本高等原因,其在移动氢源方面的应用并不具有优势。光催化和光电催化均是直接利用太阳能为能量来源,通过光照激发半导体光催化剂生成光生电子和空穴,利用光生电子还原水生成氢气。相较于光催化制氢,光电催化制氢通过外电路能够有效地分离和转移光生电子和空穴,降低了电子和空穴复合的概率,提高了制氢效率。光催化和光电催化体系中由于光吸收受限、光生电荷分离传输缓慢以及限速步骤的析氧反应缓慢等问题,其制氢效率和能量转换效率均很低,同时太阳能是间歇不稳定的,因此光催化和光电催化制氢在移动氢源方面的应用受到限制[7-12]。鉴于电催化的高成本和光催化/光电催化的制氢效率低等问题,选择了催化新型化学储氢材料进行制氢的研究。
目前,主要的化学储氢材料有氨硼烷(NH3BH3)、硼氢化钠(NaBH4)、水合肼(N2H4·H2O)等,相较于NH3BH3和NaBH4的高成本和制氢反应的副产物回收困难等特点,水合肼作为一种新型的液态化学储氢材料[13-14]具有以下优点:首先水合肼的物理化学性质稳定,生产规模大,材料价廉易得[15];其次储氢容量高(8%,质量分数),可以兼容使用现有汽、柴油的储运和加注等基础设施;最后产物环境友好,制氢反应不产生固体副产物,不产生毒害PEMFCs 的碳化物,制氢装置简单且反应条件温和可控,具备在线储/制氢一体化的技术特征,因此水合肼在车载/便携式移动氢源方面的应用潜力巨大[16]。水合肼催化制氢为非均相催化反应,固态的催化剂和液态的水合肼易于分离,水合肼制氢系统可以通过控制催化剂和水合肼溶液的接触和分离实现反应的启动和停止,制氢速率可以通过控制水合肼溶液的加入速度来进行调整。水合肼的催化分解反应和肼(N2H4)一样按如下两条竞争路径进行。完全分解:

从热力学的角度来看,式(2)的Gibbs 自由能低于式(1),即水合肼不完全分解生成氨气的反应是一个热力学更有利的过程。但不完全分解过程所产的氨气会使Nafion 膜及燃料电池的催化剂中毒[17-18],若想将水合肼作为氢源,应抑制不完全分解反应避免副产物氨气的产生,使其完全分解为氮气和氢气。水合肼制氢技术的关键在于设计和制备合适的催化剂促使水合肼快速地完全分解生成N2和H2(即H2选择性达到100%)。Singh等[19]率先采用共还原法制备出单金属纳米催化剂用于水合肼的催化制氢,实验发现在水溶液中Cu、Ni、Fe、Pt 和Pd的H2选择性为0,而Co、Ru、Ir 的H2选择性约为7%,Rh 的H2选择性最高(43.8%),由此开展了过渡金属纳米催化剂催化水合肼制氢的实验和理论计算研究。
Singh 等[20-23]制备出的Fe基、Co 基、Cu 基和Ni基纳米合金催化剂在常温无碱性助剂的情况下对水合肼的催化性能各不相同,其中Fe基、Co基和Cu基贵金属(Rh、Pt、Ir、Pd)催化剂几乎无活性,而大部分的Ni 基贵金属催化剂的H2选择性能够达到100%,由此可见催化剂的元素组成对催化性能具有决定性的作用。但通过以下改性策略可以有效地提升催化性能:一是通过载体负载和改变催化剂的形貌来增加活性位点;二是通过合金效应和载体效应来提高活性位点的本征活性,催化剂的本征活性可以通过调控活性位点的几何结构和电子结构来改变[24]。
实验发现Ni 基贵金属纳米催化剂的催化性能特别优异,其催化活性远远高于非贵金属催化剂,但由于贵金属成本高昂、资源稀缺限制了其商业化应用,目前主要从降低贵金属用量和开发非贵金属及非金属催化剂来降低成本,但非金属催化剂的研制主要局限在理论计算预测方面。纳米催化材料的比表面积巨大,其表面原子有着丰富的悬空键和不饱和键,能够产生大量的配位不饱和活性位点,改善了N2H4和反应中间体在催化剂表面的吸附和脱附性能,从而有利于提高催化性能。但由于纳米颗粒表面能高易于团聚和烧结,其稳定性较差易于失活[25-26],目前主要通过金属或非金属掺杂和载体负载来调节纳米颗粒与载体的吸附强度,减缓纳米颗粒的团聚和烧结来增强催化剂的稳定性[27]。
关于水合肼催化分解制氢的实验研究已经取得了较大的发展,积累了大量表观实验性能的数据和经验,提出了合金化、负载化和形貌结构控制等提升催化性能的催化剂制备策略,同时水合肼催化制氢的研究已经逐步从表观实验研究深入到从原子和分子层次还原催化材料和性能之间的构效关系的机理研究。理论研究目前主要是采用密度泛函理论(density function theory,DFT)计算N2H4在不同的催化剂上的吸附性能,同时结合过渡态理论来确定催化分解反应的中间态、过渡态和反应路径。本文主要从催化剂的合金化、负载化和形貌结构调控等因素和催化性能之间的构效关系进行综述,并结合目前的理论计算结果总结水合肼催化分解的相关机理研究结果,以期为实现机理指导下催化剂的结构精准设计奠定基础。
1 水合肼催化制氢的机理研究进展
催化反应主要在催化剂的表界面发生,催化剂表面原子的几何结构、电子结构和配位环境决定着催化性能,研究表界面的催化反应机理有助于阐明能够有效催化水合肼分解的活性位点的本质,从热力学和动力学上揭示活性位点上N—N 键和N—H键的活化以及反应中间体形成和转化的反应机制,实现原子级别上活性位点的几何结构、电子结构和缺陷结构的可控合成和分布。目前表界面催化理论计算研究多采用周期性晶体结构模型,主要有团簇模型(cluster model)和超元胞模型(slab model),由于团簇模型难于精确描述由结构松弛和应力效应等引起的计算结果误差,故多采用超元胞模型来模拟周期性晶体结构的催化剂表面[28]。
非均相催化的三个关键步骤:首先是反应物吸附在固体催化剂表界面的活性中心上;其次是发生表界面反应,断裂旧键生成新键形成产物;最后是产物脱附,催化活性中心再生。吸附可分为物理吸附和化学吸附,化学吸附是发生催化反应的基础,物理吸附由分子间作用力(van der Walls force)所产生,作用力较弱因而不会发生催化反应,化学吸附由化学键力所产生,能够有效地改变催化剂表界面原子的电子结构,导致吸附质和吸附剂表面原子的电子重排,从而发生化学键的断裂或形成。理论计算表明H2O 对N2H4在催化剂表面原子的吸附和分解影响很小,可以忽略[29],目前关于肼催化分解相关理论研究的文献报道多集中于N2H4在不同催化剂表面原子上的吸附性能及分解路径研究。
活性位点的本征活性遵从Sabatier 原理,通过评估活性位点与反应物和中间体之间的吸附强度来预测催化剂的本征活性,理想的活性位点既能牢固吸附反应物使反应分子发生活化,又不能吸附太强以致产物无法脱附,处于适中的强度时催化性能最优,即催化活性和吸附能之间呈现出一种火山型曲线的关系[30]。研究N2H4在不同的催化剂表面的吸附性能即可预测催化活性,故初期水合肼催化制氢的机理研究主要是围绕N2H4在催化剂表面的吸附过程展开。催化剂的H2选择性主要由N2H4在催化剂表面上的分解路径决定,而分解路径由肼分子中N—N 键、N—H 键的断键顺序决定,因此肼在催化剂表面分解路径的理论研究主要是结合密度泛函理论和过渡态理论计算N—N 键、N—H 键的断键能垒。
1.1 肼在催化剂表面的吸附研究
肼分子(NH2—NH2)由两个NH2基团通过单键(σ键)连接形成,其分子轨道(MO)中共有14 个价电子,NH2基团中的中心原子N 用1 个s 轨道和3 个p轨道不等性杂化形成4 个sp3杂化轨道,其中1 个杂化轨道有一对未成对电子(N 的孤电子对)。N2H4在气相中有三种稳定的构型:gauche、cis、anti。由于超共轭效应,gauche 构型的N2H4在气相中最稳定[31-33]。当N2H4吸附在催化剂表面原子上时,anti 和gauche构型的N2H4倾向于通过一个N 原子和催化剂表面原子吸附成键(即顶位吸附),而cis构型的N2H4倾向于通过两个N原子和金属原子吸附成键形成桥式吸附,此时N—N 轴平行于金属表面原子(图1),理论计算表明肼在催化剂表面原子上的不同几何结构是影响催化性能的因素之一。Daff 等[34]采用DFT 理论计算了不同晶面Cu(111)、Cu(100)和Cu(110)的结构敏感性,发现N2H4在不同晶面的吸附强度顺序为Cu(110)>Cu(100)>Cu(111),N2H4在Cu(110)的稳定吸附构型为桥式,认为不同晶面的结构区别是造成吸附性能出现差异的关键原因。Tafreshi 等[35]通过计算发现N2H4在不同晶面的吸附强度同样为Cu(110)>Cu(100)>Cu(111),而不同晶面原子的配位数大小顺序为Cu(110)<Cu(100)<Cu(111),即原子配位数越低的晶面吸附性能越强。纳米催化剂的催化性能和纳米颗粒大小及表面形貌是密切相关的,He等[28]认为团簇模型更适于模拟不同颗粒大小和表面形貌的纳米催化剂,故首次采用Rhn纳米团簇作为模型来模拟N2H4的吸附,DFT 计算结果表明Rhn纳米团簇中配位数较低的顶点原子是最受欢迎的吸附位点,桥式吸附是最稳定的吸附构型,在0.52~1.66 nm 范围内吸附性能随着纳米颗粒粒径的增大而增强,纳米团簇上的缺陷结构能够提供更强的吸附性能,即相较于周期性晶体结构表面在不规则的和缺陷的表面结构上更易于生成高效的催化活性中心。
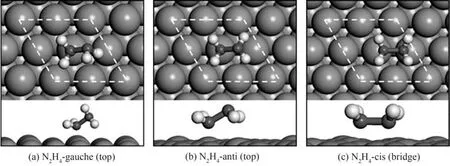
图1 肼分子在催化剂表面原子上的顶式和桥式吸附构型[29]Fig.1 The adsorption configurations of N2H4 on the surface atoms in top and bridge[29]
在纳米催化剂表面原子上的vacancy、steps、kinks、stacking faults 和terraces 等缺陷位,配位数较低从而吸附能较高,通常是高活性催化位点。Tafreshi等[35]分别计算了N2H4在Cu(111)、Cu(100)和Cu(110)晶面上配位数不同的steps、corners、vacancy等缺陷位和周期性表面的吸附性能,计算结果表明随着不同缺陷结构配位数的降低d带中心不断上移靠近费米能级,吸附性能不断提高[28,36]。He等[37]通过将单个Rh原子吸附在Rh(111)晶面上来模拟低配位数的边位和角位,计算结果同样表明低配位数的活性位点能够产生更强的吸附性能。根据键级守恒原则,若分子间力很强则相应的分子内力较弱,因此在具有更强分子间吸附力的低配位数原子上吸附的N2H4,其N2H4分子内力较弱,即更易于断裂N—N键或N—H 键[38]。通过调控纳米颗粒表面原子的缺陷结构来提高催化活性已经在实验中得到证实:Li等[39]通过调控银纳米颗粒表面原子的结构形成高密度堆垛层错(stacking faults)缺陷结构,既能有效降低银表面原子的配位数,又能产生拉伸应变,从而改善了表界面银原子对反应中间体的吸附性能,使得不具备催化活性的d10银纳米颗粒转变成高析氢催化活性的纳米材料;Barlocco 等[40]对比了未经处理缺陷很少的Graphite 和经过热处理后本征缺陷较多的碳纳米催化剂CNF-PS(carbon nanofibers pyrolytically stripped)、CNF-HHT(carbon nanofibers high heattreated)的催化性能,其中经过热处理后缺陷最多的CNF-PS 催化水合肼分解的实验性能最优,缺陷最少的Graphite的催化性能最差。
在大量的实验研究中发现,金属催化剂的合金化改性可以改善催化剂的氢气选择性和催化活性(图2),但合金化对上述性能的提升机理并不清楚。He 等[41]采用DFT 理论计算N2H4在不同掺杂比的Ni基合金表面的吸附能,计算结果表明合金的不同组成比例能够显著地影响N2H4的吸附稳定性,其理论吸附性能最优的合金组成比例和实验结果一致,同时发现N2H4在金属Ni 表面的吸附性能会随着覆盖度的改变而改变,覆盖度较小时桥式吸附比较稳定,覆盖度较大时顶位吸附是最稳定的。Shi等[26]通过DFT 理论计算发现N2H4在组成比例分别为Ni4W和Ni17W3的合金催化剂表面上最稳定的吸附构型是顶位吸附,N2H4在Ni4W 表面原子上的吸附能高于在Ni17W3上的,这和实验中Ni4W 的催化性能优于Ni17W3的测试结果一致。
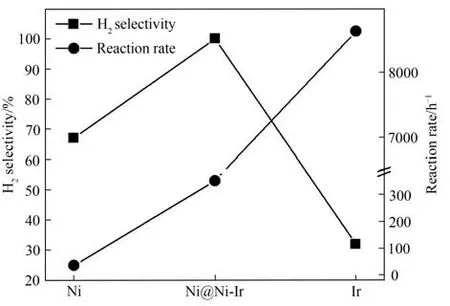
图2 金属Ni、金属Ir和合金Ni@Ni-Ir催化剂的催化性能对比[38]Fig.2 Comparison of catalytic performance of metal Ni,metal Ir and alloy Ni@Ni-Ir catalysts[38]
Cross 等[42]通过理论计算发现合金化的NiZn 双金属相较于单金属Ni 和Zn 的d 带中心更加靠近费米能级,因此其对N2H4的吸附性能也更加优异。计算发现N2H4在Ni(111)、Zn(001)、NiZn(111)、NiZn(110)、NiZn(100)晶面的吸附能分别是-1.97、-1.15、-2.71、-2.08、-2.11 eV,双 金属NiZn(111)、(110)、(100)对N2H4的吸附强度明显高于单金属Ni(111)和Zn(001)。Ni 的d带中心值在Ni(111)、NiZn(111)、NiZn(110)、NiZn(100)晶面分别为-1.65、-0.91、-1.13、-0.99 eV,Zn 的d 带中心值在Zn (001)、NiZn (111)、NiZn (110)、NiZn(100)晶面分别为-7.14、-6.51、-6.74、-6.60 eV,即合金化双金属NiZn 的d 带中心相较于单金属均出现了上移更加靠近0 eV 处的费米能级(图3)。在吸附过程中N2H4中N 的孤对电子(N-p)轨道和金属的d轨道相互作用形成成键轨道和反键轨道,根据d带中心理论当d带中心靠近费米能级时推高了反键轨道的能级,从而电子不易于占据反键轨道,因此双金属NiZn的吸附更加稳定[36]。
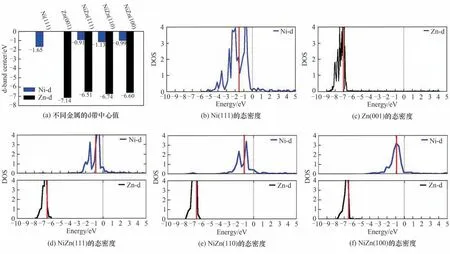
图3 Ni(111)、Zn(001)、NiZn(111)、NiZn(110)和NiZn(100)的d带中心值和电子态密度图[42]Fig.3 Calculated d-band center and the surface DOS for the Ni(111),Zn(001),NiZn(111),NiZn(110),and NiZn(100)[42]
以上均为N2H4在过渡金属原子表面的化学吸附,而关于N2H4在非金属表面的吸附研究较少。Zeng 等[43]发现N2H4在非金属石墨烯表面为物理吸附,这和实验中石墨烯对N2H4的分解反应无活性的结果一致。Esrafili等[44]研究了N2H4在半导体材料碳化硅纳米管(SiCNTs)上的吸附情况,研究发现N2H4倾向于通过一个N原子顶位吸附于荷正电的Si原子上,SiC中Si原子的杂化轨道和N2H4中N原子的轨道同为sp3杂化轨道,从而两者的轨道之间能够最大程度重叠,利于N2H4的吸附。Zheng 等[33]对比研究了N2H4在具有类石墨烯平面结构的金属碳化物XC3(X=Al,Ge)和非金属碳化物XC3(X=B,N,Si)纳米片上的吸附性能,理论计算表明吸附能以g-NC3<a-GeC3<a-SiC3<c-BC3<c-AlC3(g、a、c 分别代表gauche、anti、cis 构型)顺序递增。在NC3纳米片上N2H4的吸附是物理吸附,N2H4在其余四种XC3上为化学吸附,根据XC3中X 原子的Mulliken 电荷分布结果可知,XC3中只有N原子的Mulliken电荷为负,其余四种为正,因此N2H4在NC3上表现出物理吸附。由于SiC3在N2H4分子吸附前后均保持半金属性,因此预测非金属的SiC3能够作为N2H4分解的有效催化剂。
从政党和国家两个层面来看,意识形态建构的过程都与知识分子有必然关系。从政党意识形态宣导的过程来看,最初的“受众”局限于少数知识分子,其开发和传播也依赖于知识分子;从政党意识形态国家化的过程看,最重要的环节亦是对知识阶层的吸引。1929年,胡适在《新月》上发表文章批评国民党,即指出:“现在国民党所以大失人心,一半固然是因为政治上的设施不能满足人民的期望,一半却是因为思想的僵化不能吸引前进的思想界的同情。前进的思想界的同情完全失掉之日,便是国民党油干灯草尽之时。”瑓瑦把思想界的同情和国民党的存亡联系在一起,颇具象征意义。
1.2 肼在催化剂表面分解路径研究
N—N键的断键能垒为286 kJ/mol,N—H键的平均断键能垒为360 kJ/mol,从热力学角度分析N—N键更易于断裂。结合目前的实验和理论研究结果推测,肼的分解路径可能有三种:①分子内反应,N—H 键优先断裂,N2H4脱氢依次生成H2和N2,N2来自同一个N2H4分子,未经过N—N 键断裂;②分子间反应,N2H4中N—N 键优先断裂成NH2(a)自由基(a 代表吸附态),然后NH2依次夺取邻近N2Hx(x=1~4)中的H 原子生成N2和NH3;③分子间反应,N—N 键优先断裂生成NH2(a),然后NH2(a)依次脱氢生成N(a)和H(a),最后复合生成N2和H2。但由于N2是叁键结构,通过N(a)原子复合生成N2的能垒非常高,因此通过N—N键优先断裂的反应路径③生成N2和H2的反应过程在温和的反应条件下不可能实现,这也在以下实验中得到了证实。Maurel 等[45]采用同位素15N标记法研究了N2H4在氧化铝负载的不同过渡金属催化剂上的分解过程,实验表明N2H4在过渡金属Ru、Co、Rh、Ir、Ni、Pd、Pt、Cu 上催化分解所产生的N2均来自同一个N2H4分子,即水合肼催化分解所产N2并不是通过N—N 键断裂形成的,催化反应中优先发生断裂的是N—H 键。N2H4在Ni(100)晶面的热分解实验确认了中间体N2H2的存在[46],N2H4在Ni-Pt/CeO2[47]上的分解实验确认了反应中间体N2H3、N2H2、N2H的存在。据此推测N2H4最有可能的分解反应路径是①分子内反应和②分子间反应,其反应路径如下:

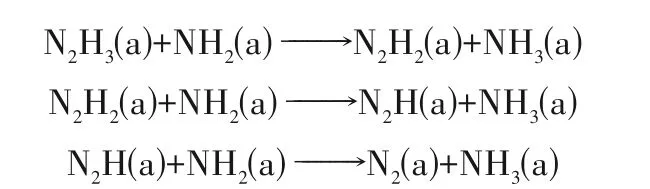
N2H4在单金属Rh(111)晶面[48]和Ir(111)晶面[49]的分解情况类似,优先断裂能垒较低的N—N 键生成NH2,然后NH2夺取邻近N2Hx(x=1~4)中的H 原子,即易于发生②分子间反应生成N2和NH3。计算结果表明反应中间体NH2能够有效地活化N—H 键,因此NH2易于夺取N2Hx(x=1~4)中的H 原子生成NH3,同时NH2辅助脱氢过程中N—N键逐渐变强,其键长按N2H4>N2H3>N2H2>N2H>N2的顺序逐渐变短,即反应中间体NH2加强了N—N键。Yin等[29]计算了N2H4在Ni(111)晶面的分解反应过程,发现表面覆盖度可以改变N2H4的分解路径,表面覆盖度为0.11 ML 时,肼分子内的N—N 键优先断裂形成中间体NH2,然后NH2通过分子间反应夺取邻近N2Hx(x=1~4)中的H 原子生成N2和NH3,当覆盖度为0.25 ML时,N—N键和N—H键的断裂是竞争性的,即①分子内反应和②分子间反应同时进行,产物有H2、N2和NH3,H2选择性为50%左右,这和实验结果一致。McKay 等[50]同样发现N2H4在Fe(211)晶面上N—N 键和N—H 键的断键能垒相差不多,能够同时进行分子内反应①和分子间反应②(竞争性反应)。Zhang 等[51]通过计算发现了N2H4在MoC、Ir、Mo、Ru、Rh、Ni、Co 上的吸附性能和活化能垒之间的线性关系,即N2H4在不同的过渡金属上的吸附性能越强,N—N 键和N—H 键的活化能垒越低,但总体N—N 键的活化能垒高于N—H 键的活化能垒,推测这可能是单金属纳米催化剂H2选择性较低的原因,通过OH 和N2H4在Ni(111)晶面的共吸附计算发现在OH 共吸附后N—H 键的断键能垒降低了29%,由此可以解释实验中碱性助剂添加后H2选择性和活性出现提升的原因。
N2H4在非金属SiCNTs (silicon-carbide nanotubes)表面上的分解路径和过渡金属上的完全不同[44],计算结果表明N2H4在SiCNTs 上断裂N—N 键生成NH2的能垒较高,而通过断裂N—H键不断脱氢生成N2H2的能垒较低,反应易于进行,但N2H2继续脱氢生成N2和H2的能垒较高,不利于生成H2。Zheng 等[33]采用DFT 理论计算了N2H4在g-SiC3(掺杂有Si 原子的二维石墨烯材料)上的分解过程,发现g-SiC3是一种零带隙半金属性质的材料,具有类似于金属的性质,其最优的反应路径和金属上的一样,同为NH2辅助的分子间反应②,通过对比NH2共吸附前后的N—N、N—H键长和N2H4在g-SiC3上的吸附能发现,NH2共吸附后N2H4的吸附变强、N—N 键变短、N—H 键拉长,因此利于发生分子间反应②。由于C 的电负性大于Si,从而C荷负电Si荷正电,荷正电的Si是最受欢迎的活性位点。Genç 等[52]将单个Ni 原子嵌入蜂窝式层状结构的石墨烯的六角环中(Ni1/C31)来模拟单原子催化剂,计算结果表明由于N 的2p 和Ni 的3d 轨道之间的相互作用,N—N 键相较于N—H 键优先发生断裂形成NH2,然后NH2依次夺取N2Hx(x=1~4)中的H原子发生分子间反应②。这些非金属的理论计算研究有助于开发和预测高效廉价的非金属催化剂。
目前关于N2H4分解的机理有很多争议,以上N2H4分解路径分析主要基于热力学上的断键能垒,但是全面分析N2H4的催化分解过程需要同时考虑热力学和动力学过程。同时关于合金化、载体的负载和碱性助剂对分解路径影响的机制还缺乏相关深入的研究,因此关于催化剂的结构和水合肼催化分解性能之间的构效关系解析还需要进一步结合理论计算和先进的实验表征来进行深入的研究。
2 水合肼制氢催化剂改性制备进展
2.1 催化剂的合金化改性
单金属在高温下易于团聚、烧结稳定性不高,其表面易于被空气氧化形成钝化物,导致催化性能下降,而当Ni 和另一种金属或类金属形成合金后,能够显著地提高催化剂的稳定性。金属纳米颗粒形成合金后其表面原子排列方式和电子状态都会发生较大的变化,导致催化剂纳米颗粒表面原子的几何结构和电子结构的改变,从而能够有效地调整催化剂的吸附性能,稳定反应中间体,降低N—H 键的断键能垒[39,53]。催化剂表面原子在合金化过程中会产生晶格膨胀,这些表面原子的几何结构改变会产生应变效应(strain effect)[30],同时双金属之间的轨道杂化改变了态密度和d 带中心位置,从而产生配体效应(ligand effect),因此催化剂的本征活性能够得以有效的提升[54-55]。
Singh 等制备了不同的双金属NPs(nanoparticles,纳米颗粒)催化剂:Ni/Pd[20]、Rh/Ni[21]、Ni/Ir[22]和Ni/Pt[23],测试发现采用共还原法制备的上述双金属NPs的催化性能均远远高于对照组的单金属催化剂和物理混合的双金属催化剂。通过表征发现上述双金属NPs 形成了合金,其表面原子的几何结构和电子结构发生了显著变化,因此其催化性能相较于对照组得到了显著提升[56]。贵金属Pt、Rh、Ir、Pd 的原子半径大于金属Ni 原子,根据布拉格原理,将其掺杂进Ni 原子后晶格会出现膨胀,因此在X 射线衍射(X-ray diffractometer,XRD)表征中可以观察到合金NPs 的衍射峰位置向低衍射角偏移,由此确认合金的形成,同时通过扩展X 射线吸收精细结构谱(extended X-ray absorption fine structure, EXAFS)的检测也能发现杂金属键的形成。通过X射线光电子能谱(X-ray photoelectron spectroscopy, XPS)表征中合金催化剂表面原子轨道谱峰的结合能位置偏移,确认双金属表面的电子状态在合金形成过程中也发生了显著变化。
如图4所示,Qiu等[38]为了在催化剂表面形成Ni-Ir合金降低贵金属Ir的用量,首先采用胶体溶液燃烧法(colloidal solution combustion synthesis,CSCS)合成Ni/meso-CeO2,然后采用电镀法使得Ni/meso-CeO2表面的金属Ni被Ir取代合成Ni@Ir/meso-CeO2,最后再进行煅烧处理使得Ni@Ir/meso-CeO2表面的金属Ni和Ir 形成合金,即制得表面合金化的Ni@Ni-Ir/meso-CeO2高效催化剂。实验结果表明表面合金化的Ni@Ni-Ir/meso-CeO2催化剂的催化性能高于非贵金属Ni 但低于贵金属Ir,这和DFT 理论计算的吸附结果一致,N2H4的吸附强度顺序是Ni<Ni@Ni-Ir<Ir。Zhong 等[57]结合共沉淀法和电镀法制备出合金催化剂Ni@Ni-Pt/La2O3,其H2选择性为100%、反应时间为10 min,相较于单金属Pt/La2O3完全无活性和Ni/La2O3微弱的催化性能,NiPt 的合金化是催化剂Ni@Ni-Pt/La2O3的催化性能得到大幅提升的关键因素。在俄歇电子能谱(auger electron spectroscopy,AES)的检测中发现由于Ni和Pt的合金化,Ni@Ni-Pt/La2O3中位于费米能级以下的Ni 原子的d 带带宽相较于Ni/La2O3变窄,因此其d 带中心位置会出现上移从而更加靠近费米能级,N2H4中N原子和Ni@Ni-Pt/La2O3中Ni原子吸附形成的反键轨道能级变高,反键轨道能级高不利于电子占据,因此N2H4在Ni@Ni-Pt/La2O3上的吸附更加稳定,即合金化的Ni@Ni-Pt/La2O3的催化性能更加优异,这和d带中心理论一致。

图4 催化剂Ni@Ni-Ir/meso-CeO2的制备示意图[38]Fig.4 Schematic illustration of the synthesis of Ni@Ni-Ir/meso-CeO2 catalyst[38]
研究发现催化剂的H2选择性强烈地依赖于金属组成比例,如图5所示,Rh4Ni的H2选择性为100%而RhNi4仅为71%[21],Ni0.93Pt0.07的H2选择性为100%而Ni0.26Pt0.74仅为10%[23]。根据Sabatier 原理,存在一个最佳的金属组成比例,使得催化剂表面的活性组分对N2H4的吸附既不过强也不过弱,此时的催化性能最优异[58]。由于常温下H2的解吸过程可能是水合肼分解反应的限速步骤,同时N2在催化剂表面的过强吸附会导致催化剂的失活[47],而金属催化剂在合金化过程中表面原子的Metal-H、Metal-N 键的强度会被显著削弱,从而反应中间体和反应产物(H2、N2)的解吸变得更加容易,因此催化剂的转换频率(turnover frequency,TOF)和H2选择性均能得到有效提高。He 等也发现由于Ni-Pt[59]、Ni-Ir[60]的合金化,催化剂表面原子的Metal-H、Metal-N 键的强度被显著减弱,提升了催化性能。
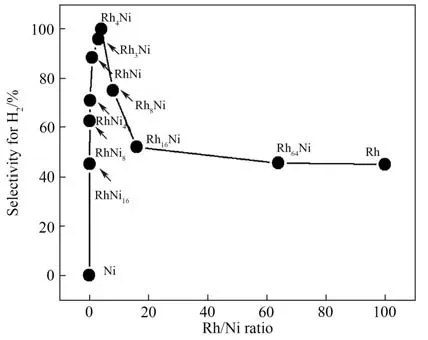
图5 摩尔比不同的RhNi催化剂催化分解水合肼的H2选择性[21]Fig.5 Selectivity for hydrogen generation from hydrous hydrazine catalyzed by different Rh/Ni molar ratio[21]
因贵金属催化剂资源有限和价格昂贵,其大规模的商业应用受到限制,由此展开了非贵金属和非金属掺杂制备纳米合金催化剂的研究,如Co/ZnO@NiFe2O4[61]、NiCu/CeO2[62]、NiMo/TiO2[63]、NiFePd[64]、Ni10Mo/Ni-Mo-O/Ni foam[65]、Rh-Ni-B[66]、RhP/rGO[67]、NiPtP/rGO[68]等。
2.2 催化剂的形貌和结构调控
在非均相催化反应中纳米催化剂表界面的成分和结构决定着催化性能,催化剂表面上经常暴露着不同的晶面,晶面不同则表面原子排布不同,因此其催化性能也不同。Wang 等[69]通过合成核壳结构的纳米催化剂来调控催化剂的表界面成分和结构,采用晶种生长法合成Cu 为核FeNi 为壳的Cu@FeNi 催化剂(粒径约为8.5 nm),使活性成分FeNi 合金充分暴露在催化剂表面而不是体相中,这样既能提供更多的活性位点又能通过改善催化剂表界面结构来提高催化剂的本征活性,因此其催化性能相较于普通合金催化剂得到了大幅提升。Zhong 等[57]采用电化学置换法将微量的贵金属元素Pt 引入到Ni/La2O3催化剂表面,通过热处理使Ni-Pt合金化,成功制备出催化性能优异的核壳结构的Ni@Ni-Pt/La2O3催化剂,其H2选择性为100%,反应速率为312 h-1(50℃,碱性环境)。Oliaee 等[70]采用固态化学法合成特定形貌的八面体Pt-Ni/C 纳米催化剂,使其暴露的晶面均为(111)晶面,通过实验发现单晶面(111)催化剂的H2选择性(100%)高于各种晶面混杂的球形催化剂(75%),但是单晶面(111)催化剂的TOF低于球形催化剂。
Yang 等[71]采用液相等离子体技术合成具有独特一维纳米结构的Co-B-N-H 纳米线催化剂,此外由于N 和H 原子的掺杂不仅增加了强碱性位,而且有效地削弱了反应中间体和Co 表面原子的吸附性能,因此其催化活性和稳定性均很优异,在无碱性助剂条件下约17 min 完全分解水合肼,H2选择性达到100%,活化能垒为(39.9 ± 1.5) kJ/mol。Fu 等[72]采用静电纺丝和真空热处理法制备出纤维状的Ni 纳米催化剂(比表面积132 m2/g),其H2选择性为100%,TOF 为6.9 h-1(60℃,碱性溶液),而颗粒状的H2选择性仅为30%。Wang 等[73]采用真空热处理共还原法制备出纤维状的Ni-Cu 纳米合金,其催化性能相较于颗粒状的Ni-Cu合金更加优异。
催化性能不仅可以通过改变催化剂本身的形貌结构,而且可以通过改变催化剂载体的形貌结构来调控。有报道指出将CoPt 纳米合金颗粒负载在形貌各异的纳米片、纳米球、纳米棒和纳米带状载体La(OH)3上,CoPt/La(OH)3的催化性能会随着载体La(OH)3形貌的改变而改变(图6)[74],其中负载在比表面积最大的La(OH)3纳米片上的CoPt 纳米合金催化性能最优,TOF高达2400 h-1(50℃)。Prabu等[75]采用Al(OH)3纳米片作为载体制备NiPt/Al(OH)3催化剂,25℃下TOF 就达到了1280 h-1。Wu 等[76]通过动态控制共沉淀-还原工艺和煅烧处理过程成功地将NiCo 合金负载在形貌不同的层状纳米片、海胆状、纳米颗粒状的NiO-CoOx载体上(比表面积分别为198.4、256.9、82.21 m2/g),并对比分析了各载体对催化性能的影响(表1),结果表明催化性能最优的是层状纳米片负载的NiCo 合金,而不是比表面积最大的海胆状载体负载的NiCo 合金,认为原因如下:一是层状纳米片的碱性位点多于海胆状;二是层状纳米片NiO-CoOx上的配位不饱和O2-有很多孤对电子,使得N2H4分子更易于与其形成氢键,削弱了N—H键的强度,同时在活性中心NiCo 的活化作用下能够有效地促进N2H4分解反应中限速步骤N—H 键的断裂,从而H2选择性和催化活性得以提升。
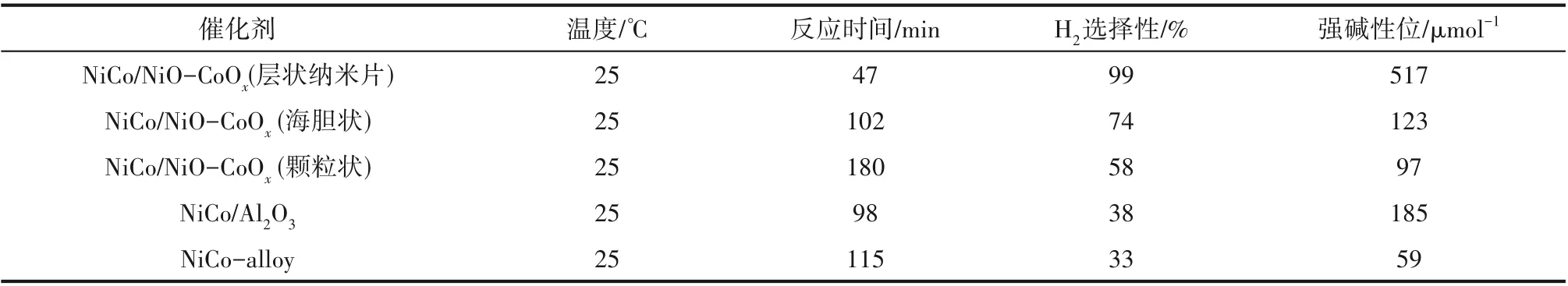
表1 不同形貌的NiCo/NiO-CoOx、NiCo/Al2O3 和NiCo NPs的催化性能和碱性位Table 1 The catalytic performance and basic properties of NiCo/NiO-CoOx,NiCo/Al2O3,and NiCo NPs with different morphologies
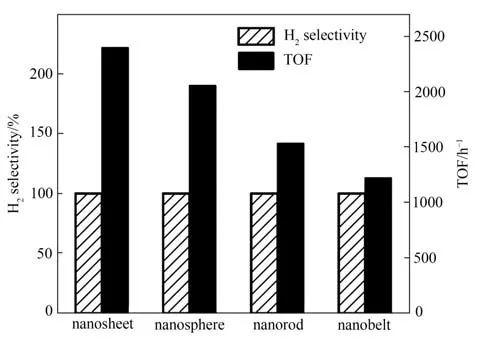
图6 载体形貌不同的CoPt/La(OH)3催化分解水合肼的H2选择性和TOF对比(50℃,cNaOH=3.5 mol/L,nCoPt/nN2H4·H2O=0.05)Fig.6 The H2 selectivity and the corresponding TOF values of dehydrogenation of hydrous hydrazine over CoPt/La(OH)3with different shape of La(OH)3(50℃,cNaOH=3.5 mol/L,nCoPt/nN2H4·H2O=0.05)
2.3 强碱助剂的促进作用


虽然使用强碱性助剂可以提高催化剂的性能,但在反应体系中加入强碱性助剂容易引起反应容器和输送设备的腐蚀,而将金属NPs 负载在碱性载体上既可以提升催化性能又可以避免强碱性溶液的腐蚀作用。He 等[85]采用强碱性Al2O3作为载体制备出Ni-Al2O3-HT 催化剂,Gao 等[86]将强碱性氧化物MgO 作为载体制备出NiFe-alloy/MgO 催化剂,其氢气选择性均超过了90%。
2.4 催化剂载体的影响
金属纳米颗粒催化剂表面能高、稳定性低,易于烧结团聚而失活,同时分离回收困难,因此其在工业催化中的应用受到了限制。将金属纳米颗粒高度分散在载体上能够有效增加催化剂的比表面积和分散度,提供更多的活性位点,同时载体和金属之间强相互作用(strong metal-support interaction,SMSI)能够有效地改变催化剂表面原子的几何和电子结构,调控活性金属原子的配位环境及金属和载体之间的电荷分布,改变反应中间体的吸附能力,提高纳米催化剂的稳定性和本征活性[27,30]。目前报道的主要载体有碳基载体、金属氧化物、金属有机骨架和其他复合载体等,不同负载型催化剂的催化性能见表2。
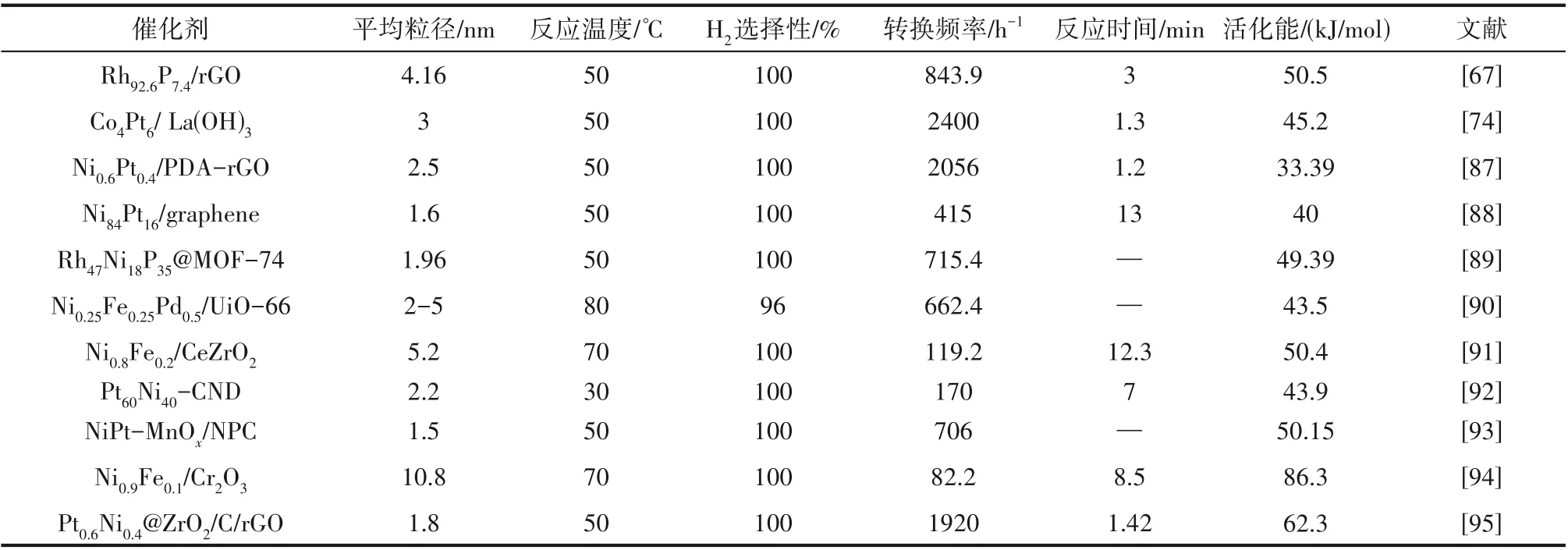
表2 不同载体负载的金属催化剂在碱性条件下催化分解水合肼产氢的性能比较Table 2 The comparison of catalytic performance for hydrous hydrazine decomposition over metal catalyst with different supports
2.4.1 碳基载体 石墨烯(graphene)及其衍生物是一种单原子厚的二维纳米碳材料,具有以下特点:比表面积大,表面电子转移能力强,表面结构和性质易于调整,热/电导率良好,柔韧性好。Wang 等[80]采用石墨烯作载体制备出平均粒径为5 nm 的RhNi@graphene 催化剂,而RhNi NPs 的平均粒径26 nm,实验中RhNi@graphene 的催化性能也远远高于RhNi NPs,由此可见石墨烯载体能够有效地提升催化剂的分散性,从而提高催化性能。Song 等[87]进一步采用对苯二胺(PDA)对氧化石墨烯(rGO)载体进行碱化制备出Ni0.6Pt0.4/PDA-rGO 催化剂,由于PDA 中的氨基有利于NiPt NPs 负载在rGO 上,同时二胺碱化的rGO 促进电子从载体转移到金属活性中心,从而形成富电子状态的NiPt NPs,因此相较于未加PDA 的Ni0.6Pt0.4/rGO 催化剂,其TOF 大幅提升。
类石墨相氮化碳(g-C3N4)作为一种新型的环境友好型非金属催化材料,有着极高的比表面积,良好的热稳定性和化学稳定性。其原子尺度的超薄层状结构可以提供高比例的配位不饱和位点,利于N2H4的吸附从而提高催化性能。Xu等[96]采用g-C3N4作为载体制备出平均粒径为3.2 nm 的NiPt/g-C3N4催化剂,其中Ni37Pt63/g-C3N4在50℃的碱性溶液中能完全分解水合肼,TOF 达到570 h-1。NiPt/g-C3N4优异的催化性能可以归因于NiPt 合金化效应以及活性组分NiPt NPs和载体g-C3N4之间的协同效应。随后该课题组采用二维平面结构的类石墨相氮化碳纳米片(g-C3N4NSs)作为载体制备出平均粒径为2.8 nm的NiPt/g-C3N4NSs 催 化 剂[97],由 于g-C3N4NSs 载 体独特的二维平面结构,NiPt/g-C3N4NSs 的催化活性较NiPt/g-C3N4又得到了进一步的提升。Qiu 等[98]将Ni2+/PtCl2-6-PDA(聚多巴胺)包覆于硬模板SiO2表面,然后通过还原退火处理形成表面合金,最后脱除模板SiO2,即制备出层状多孔结构的NiPt/NC 催化剂(图7)。由于载体独特的层状多孔结构提高了催化剂的分散性,以及通过还原退火处理加强了SMSI效应。此外,NiPt 的合金化改善了催化剂表面原子的结构,因此NiPt/NC的催化性能优异。
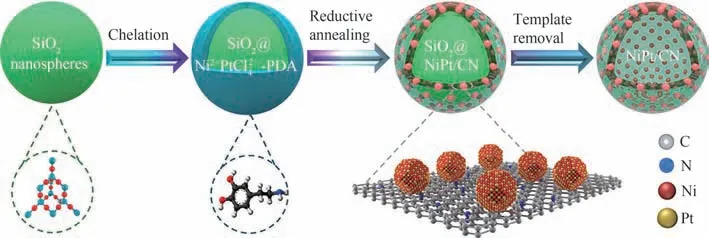
图7 催化剂NiPt/NC的制备示意图[98]Fig.7 Schematic diagram of the fabrication process of NiPt/NC nanocomposite catalyst[98]
2.4.2 金属氧化物载体 金属氧化物和载体之间存在着很强的金属-载体相互作用(SMSI),使得催化剂表面原子的电子状态发生改变,有助于催化性能的提升。Chen 等[94]采用Cr2O3为载体制备出小粒径、高比表面积的NiFe-Cr2O3催化剂,Yao 等[99]采用共还原法制备出非晶态NiPt-MoOx催化剂,以上催化剂的催化性能较镍基合金NPs 均有很大的提升。基于XPS 中结合能的偏移推测,电子从金属氧化物载体(Cr2O3和MoOx)转移到Ni 基合金中,从而Ni基合金的电子态密度增加,电子更易于从镍基合金转移到肼分子的反键轨道上,利于N—H 键的断裂。Song 等[95]发 现Pt0.6Ni0.4@C/rGO 的 催 化 性 能低于Pt0.6Ni0.4@ZrO2/C/rGO (制备流程如图8 所示),Pt0.6Ni0.4@ZrO2的催化性能低于Pt0.6Ni0.4@ZrO2/C/rGO,平均粒径为1.8 nm 的Pt0.6Ni0.4@ZrO2/C/rGO 催化性能极佳,能在50℃的碱性溶液中1.42 min 内完全分解水合肼,H2选择性为100%,TOF 达到1920 h-1。这是由于电子从ZrO2转移到PtNi NPs 上,形成富电子状态的PtNi NPs,因此催化性能得以提升。

图8 纳米催化剂PtNi NPs@ZrO2/C/rGO的制备示意图[95]Fig.8 Schematic illustration for the immobilization of PtNi NPs on the metal-organic framework templated ZrO2/C/rGO support[95]
非晶态材料具有长程无序、短程有序以及各向同性的特殊结构[100],同时非晶态合金表面有着高浓度的配位不饱和位点,因此非晶态催化剂的活性和选择性优异[101]。Wang 等[102]采用稀土化合物作为载体制备出非晶态的NiPt/Ce2O3催化剂,实验中发现常温下NiPt/Ce2O3的催化性能(TOF 为28.1 h-1)高于粒径相近的晶态NiPt NPs(TOF 为6.4 h-1)。随后该课题组在CoPt/CeOx催化分解水合肼的实验中[103]同样发现非晶态催化剂CoPt/CeOx的催化性能高于晶态CoPtNPs 催化剂,考虑稀土氧化物CeOx的掺杂破坏了NiPt 和CoPt 纳米合金的长程有序性继而形成非晶态,从而催化性能得以提升。但非晶态合金表面缺陷较多,势能相对较高,在热力学中是一种亚稳态结构,在一定的温度范围内更易于向热力学稳定的晶态转变,易晶化失活、稳定性不高,因此在性能测试实验中仅经过三次循环测试活性即出现严重的衰退,TEM 表征中也发现测试后的催化剂出现团聚现象。Liu 等[63]制备的非晶态催化剂NiMo/TiO2的活性经过十次循环测试后也出现了明显的衰减。
2.4.3 其他载体 金属有机骨架材料(metal-organic frameworks,MOFs)是一种过渡金属离子作为结点与有机配体作为连接桥通过自组装形成的具有周期性网格结构的多孔材料,其表面暴露着大量配位不饱和的金属位点,具有高孔隙率、大比表面积、拓扑结构多样性的优点。将金属纳米颗粒负载在不同系列的MOFs上(如ZIF系列、MIL系列和UiO系列),不仅可以限制金属NPs的生长和团聚,提供更多的活性位点,而且可以改善活性中心的电子转移,从而提高催化剂的活性和稳定性。Xia 等[104]采用浸渍法将NiRh合金负载在ZIF-8上,负载后的NiRh@ZIF-8比表面积高达786 m2/g,平均粒径仅为1.2 nm。催化剂NiRhNPs 的氢气选择性为76%,而负载后NiRh@ZIF-8 的氢气选择性为100%,同时转换频率也大幅提高。Liu等[105]采用简易的浸渍法将NiPt NPs负载在含有不同功能基团(—NH2、—NO2、—SO3H)的MIL-101 上,由于—NH2是电子给体,而—NO2和—SO3H是电子受体,因此NiPt/MIL-101-NH2体现出最优的催化性能,在7 min内完全分解N2H4·H2O(25℃,碱性溶液),H2选择性为100%,TOF为137 h-1。
MXene 是一种新型的二维纳米层状过渡金属碳/氮化物材料,具有类似石墨烯的电导性质和亲水性,与金属有良好的相互作用,因此作为载体的潜力巨大。Liu 等[106]采用一步湿化学方法成功地将RhNi NPs 负载在改性的MXene 上,制备出平均粒径为2.8 nm 的RhNi/MXene 催化剂,其中Rh0.8Ni0.2/MXene 的催化性能最优,H2选择性为100%,50℃时TOF 达到857 h-1。Yin 等[107]采用MnOx改性的MXene为载体制备出RhNi/MnOx-MXene NPs 催化剂,由于无定形的MnOx具有高浓度的不饱和配位点,可以加强金属NPs 和载体之间的相互作用,因此双金属载体的RhNi/MnOx-MXene NPs 的催化性能相较于RhNi/MXene NPs 得到大幅提升,H2选择性达到100%,TOF 为1101.9 h-1。同时MXene 表面的功能基团Ti—OH 和Ti—F 可以将金属NPs 很好地锚固在载体上并增加催化剂的亲水性,这些都有利于提高活性组分的分散性和催化剂的稳定性。
3 结语与展望
发展水合肼可控制氢技术的关键在于研发具有高制氢选择性、高催化活性和高稳定性的低成本催化剂。总结前人的研究发现,通过合金化改性,调控催化剂的形貌和结构,增加强碱助剂,使用载体负载等策略均能有效提高催化性能。但目前的理论研究并不能准确还原水合肼催化分解制氢的反应机理,缺乏关于不同催化剂催化活性中心的位置、几何和电子结构与催化性能之间的构效关系的理论研究,不能从原子、分子和电子层次揭示催化剂活性中心的本质及催化剂与肼分子相互作用的动态过程,水合肼分解制氢高效催化剂的设计和制备缺乏理论指导,其高效催化剂制备的实验研究需要大量重复性的制备和性能测试工作,实验费时且成本高昂。因此需要结合理论计算和实验研究来系统全面地阐明水合肼催化制氢的机理,以期实现高效催化剂特定表界面结构和活性位的多尺度精准构筑,精准调控与催化反应直接相关的功能活性中心的几何结构和电子结构,为实现机理指导下催化剂的结构精准设计奠定基础。
目前的研究结果表明负载型Ni 基催化剂性能已经很优异,但要实现将水合肼催化制氢作为PEMFCs 在线氢源的商业化应用,还需解决以下问题:一是提高催化剂的活性和稳定性,针对催化剂寿命这一关键性能开展深入的研究;二是进一步降低水合肼催化制氢的成本;三是设计和开发具有自主知识产权的基于水合肼催化制氢的燃料电池在线供氢反应系统和装置。
