新诊断2型糖尿病合并高尿酸血症患者的临床特征及肠道菌群变化
2022-08-09陈柔柔赫荣波
陈柔柔,李 娜,赫荣波,邹 婧,刘 青,张 颖,刘 煜
南京医科大学附属逸夫医院内分泌科,江苏 南京 211100
高尿酸血症(hyperuricemia,HUA)是因嘌呤代谢紊乱而使尿酸生成过多和(或)排泄减少导致的一种代谢性临床综合征[1]。HUA 与2 型糖尿病(type 2 diabetes mellitus,T2DM)、代谢综合征、高血压、心血管疾病、慢性肾病、痛风等密切相关,是这些疾病发生发展的独立危险因素[2-6]。研究表明[7]尿酸水平的升高可直接或间接导致空腹血糖水平的升高,且尿酸水平可能与T2DM 的早期发病机制密切相关。糖尿病患者常伴有肾脏血流量减少,使肾小球缺氧,并使乳酸的生成增加,进而与尿酸竞争性排泄,从而导致尿酸的排泄减少。此外,糖尿病患者中多数伴有肥胖症、脂代谢障碍、胰岛素抵抗等,均会影响尿酸代谢。因此,糖尿病与HUA 可能存在共同的发病基础。
肠道微生态作为人体中最大的微生态系统,被认为是调节宿主代谢的一个重要器官,存在于人体肠道的大量微生物被称为肠道菌群。现有研究显示[8],肠道菌群组成变化与多种疾病均具有相关性,包括HUA、糖尿病、代谢综合征、肾脏病、冠心病和肿瘤等。
本研究主要通过对不同病程的T2DM合并HUA的临床特征进行分析,并进一步分析T2DM 合并HUA的肠道菌群结果特点,探究T2DM合并HUA的发生发展是否与肠道菌群的组成改变相关,旨在为新诊断T2DM 合并HUA 的微生态治疗提供新的理论依据。
1 对象和方法
1.1 对象
本研究随机纳入2019年7月—2020年7月在南京医科大学附属逸夫医院内分泌科及健康管理中心就诊的18~80 岁患者,包括单纯新诊断T2DM 患者103例和T2DM合并HUA患者98例。T2DM合并HUA 患者中,新诊断的患者29 例,病程1~5 年的患者27例,病程>5年的患者42例。从新诊断T2DM合并HUA组及单纯新诊断T2DM组中各选取年龄及性别匹配的患者,纳入9例新诊断T2DM合并HUA的患者(DMUA组),8例单纯新诊断T2DM的患者(T2DM组),并同期收集年龄及性别匹配的8 例HUA 患者(HUA组)及8例健康志愿者(Control 组),分别采集粪便标本。并同时收集所有研究对象的一般资料、实验室检查结果。本研究方案经南京医科大学附属逸夫医院伦理委员会批准(No2017⁃SR⁃001.S2)。
入组标准:①单纯HUA患者:年龄18~80岁;参照《2013高尿酸血症和痛风治疗的中国专家共识》,正常饮食状态的非同日2次的空腹水平所检测出的血尿酸值:男性血尿酸的水平≥420 μmol/L,女性血尿酸的水平≥360 μmol/L;未用降尿酸药物。②单纯新诊断T2DM患者:年龄18~80岁;参照《中国2型糖尿病防治指南2017 年版》的糖尿病诊断标准;新诊断的2 型糖尿病患者,即12 个月内诊断为2 型糖尿病患者(WHO 标准,1999 年);未用任何药物,包括降糖及调脂药物等。③新诊断T2DM 合并HUA 患者:同时满足①和②的入组标准。④T2DM 合并HUA 患者:年龄18~80 岁;参照《中国2型糖尿病防治指南2017年版》的糖尿病诊断标准;正常饮食状态的非同日2次的空腹水平所检测出的血尿酸值:2次的血清尿酸水平男性≥420 μmol/L,女性≥360 μmol/L。T2DM 病程<1 年的患者无用药史;病史1~5 年的患者仅单用二甲双胍;病史>5年的患者为单用二甲双胍或二甲双胍联合基础胰岛素,未用降尿酸、调脂药物及利尿剂等。
健康对照组:年龄18~80 岁;血糖正常,无糖尿病家族史;正常嘌呤饮食状态下,尿酸正常;近3 个月内未服用微生态制剂或抗生素;近1 个月内无腹泻及其他胃肠道的疾病史,无胃肠道手术史;无长期服用药物史。
排除标准:近3 个月内服用微生态制剂或抗生素者;有腹泻及其他胃肠道的疾病史;长期酗酒、大量进食动物内脏及海产品等习惯的患者;严重心脑、肝肾功能障碍;1 型或其他类型糖尿病;长期使用的药物不符合入组标准。
1.2 方法
1.2.1 一般资料及样本收集
患者临床信息采集主要通过随访问卷、医院信息系统等多种途径获取患者的基本信息、临床检验结果和临床特征。采集研究对象清晨排便后的新鲜粪便标本约5 g,置于密闭无菌粪便采集盒内,分别编号。所有粪便采集盒的管壁及管帽上均标注患者的姓名、编码等基本信息(油性笔做好标记),并于30 min 内迅速储存于-80 ℃低温冰箱中,避免反复冻溶。
1.2.2 粪便16S rRNA基因测序
粪便样本基因组DNA的提取采用SDS方法,并进一步使用琼脂糖凝胶电泳法检测DNA 纯度及浓度,随后将适量的样品放置在离心管中,用无菌水稀释样品至1 ng/μL。选择所需进行测序的区域,采用带Barcode 特异性引物,16S V3⁃V4 区的引物为341F:CCTAYGGGRBGCASCAG;806R:GGAC⁃TACNNGGGTATCT AAT,进一步对细菌的16S rD⁃NA V3⁃V4区进行PCR扩增。根据PCR 产物浓度进一步进行等浓度的混样,并充分混合均匀,选取1×TAE 浓度2%琼脂糖胶电泳进行纯化PCR 的产物,主带大小选择在450~550 bp之间的序列,并予割胶回收目标条带。文库的构建采用美国Illumina 公司的TruSeq DNA PCR⁃Free Library Preparation Kit 建库试剂盒。经过Qubit 的定量及文库检测后,将构建好的符合后续测序要求的文库用NovaSeq6000进一步行上机测序。测序所获得的信息通过优化序列区分样本后进行OTU聚类和物种的分类分析。
1.2.3 OTU聚类和物种分类分析
α多样性分析:主要用于分析组内微生物群落的多样性[9]。本研究采用了chao1 指数、PD_whole_tree指数、goods_coverage指数和ACE指数进行统计。
β多样性分析:基于Weighted Unifrac 距离以及Unweighted Unifrac 距离可进行PCoA(Principal Coordinates Analysis)分析[10],选择贡献率最大的主坐标的组合作图后进行展示。若样本距离越接近,则表示物种的组成结构越相似。
OTU差异显著性分析:LDA EffectSize(LEfSe)作为发现和解释高维度的生物标识分析工具之一[11],可在组与组间寻找出组间的差异显著物种,本研究用LDA值柱状分布图及进化分支图来显示。
1.3 统计学方法
采用SPSS23.0 的统计学软件对本实验的数据进行统计分析。对正态分布计量资料的数据采用均数±标准差()描述,两组间比较用独立t检验计算,两组以上用方差分析,对于方差不齐的数据采用Wilcoxon 秩和检验,组间两两比较采用Tukey⁃HSD 检验。通过Qimme 软件分析Beta 多样性。分组样品的物种组成、群落结构进行差异显著性检验采用的是T⁃test、LEfSe、MetaStat 等统计学分析方法。P<0.05 为差异有统计学意义。
2 结果
2.1 临床特征比较
比较98 例T2DM 合并HUA 患者不同糖尿病病程(病程<1 年、1~5 年和>5 年)的临床特征,结果显示,不同病程患者的年龄、低密度脂蛋白、并发症发生率差异有统计学意义(P<0.05)。新诊断T2DM合并HUA 组(病程<1 年)比病程大于5 年组的低密度脂蛋白显著升高(P<0.05),年龄及并发症发病率显著降低(P<0.05,表1)。

表1 不同病程T2DM合并HUA的一般情况及临床资料比较分析Table 1 General clinical data of patients with T2DM complicated with HUA in different stages(n=98)
比较新诊断T2DM 合并HUA 与单纯新诊断T2DM患者的临床特征,结果显示新诊断T2DM合并HUA 组的糖化血红蛋白水平显著降低(P<0.05),而总胆固醇、甘油三酯、胰岛素、C 肽水平、血清尿酸、空腹葡萄糖、动脉硬化指数及脂肪肝发生率显著升高(P<0.05,表2)。

表2 新诊断T2DM合并HUA与单纯新诊断T2DM的比较分析Table 2 Comparison of newly diagnosed T2DM patients with HUA and newly diagnosed T2DM patients
2.2 DMUA组与T2DM组、HUA组及Control组的肠道菌群分析
2.2.1 临床资料比较
4组研究对象的年龄、性别、体重指数(BMI)、转氨酶无明显差异(P>0.05)。DMUA 组与T2DM 组、HUA 组及Control 组相比,高密度脂蛋白降低,甘油三酯显著增高(P<0.05)。DMUA 组与T2DM 组、HUA 组相比,其动脉硬化指数亦明显升高(P<0.05,表3)。

表3 研究对象的一般情况及临床资料比较Table 3 General clinical data of patients
2.2.2 基于OTU的韦恩图
4 组样本共同存在的OTU 有318 个,而特有的OTU中,T2DM为38个,HUA为33个,DMUA为32个。在4 组样本中大部分的菌落是相似的,但又同时存在各自特有的差异性菌落(图1)。

图1 OTU的韦恩图Figure 1 Venn diagram of OTU
2.2.3 α多样性分析
图2 所示的是描述组内样本的多样性曲线,即稀释曲线(rarefaction curve),该曲线直接反映出所测序的数据量的合理性,并间接反映出其样本物种的丰富度,曲线趋于平坦时,表示所测序的数据量渐进合理。4组粪便样本细菌稀释曲线末端相对平坦,检测样本可满足后续实验。

图2 OTU指数稀释曲线Figure 2 OTU exponential dilution curve
α多样性分析结果显示,α多样性指数中chao1指数、ACE 指数、PD_whole_tree 指数及goods_coverage指数比较,DMUA组、T2DM组及HUA组与Control组相比均显著降低(P<0.01,图3),但DMUA 组与T2DM组及HUA组相比,α多样性指数均无明显差异。
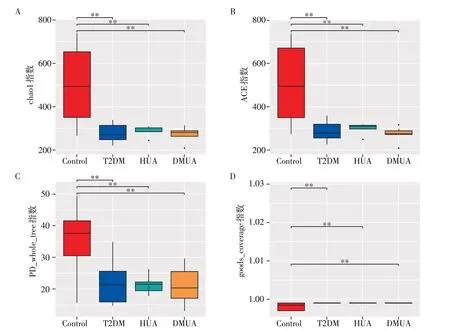
图3 各组肠道菌群α多样性指数箱式图Figure 3 Box diagram of α diversity index of intestinal flora in each group
2.2.4 β多样性分析
基于Unweighted Unifrac 距离分析显示,DMUA组较Control 组和HUA组菌群结构存在显著差异(表4);另外,基于Weighted Unifrac距离分析结果表明,DMUA组较T2DM组(P=0.002 4)和HUA组(P=0.01)均有明显差异(表4)。图4 的PCoA 结果表明,4组样本的离散较好,推断组间菌群结构存在显著差异。

图4 基于weight unifrac距离的PCoAFigure 4 PCoA based on weight Unifrac distance
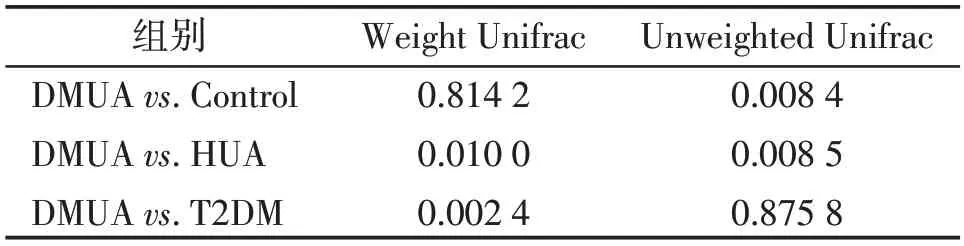
表4 用Weighted Unifrac 及Unweighted Unifrac 分析β多样性差异Table 4 Weighted Unifrac and Unweighted Unifrac were used to analyze β diversity(P值)
2.2.5 肠道菌群物种组成分析
图5A 显示的是各组研究对象在门分类水平的相对分布柱状图。各组丰度排名前4 的物种,Con⁃trol组是厚壁菌门(68.25%)、拟杆菌门(22.89%)、变形杆菌门(4.65%)、放线菌门(3.26%),T2DM组是厚壁菌门(49.98%)、拟杆菌门(38.58%)、变形杆菌门(7.68%)、放线菌门(3.30%),HUA 组是拟杆菌门(46.81%)、厚壁菌门(33.28%)、变形杆菌门(12.99%)、梭杆菌门(4.74%),DMUA 组是厚壁菌门(53.55%)、拟杆菌门(29.46%)、变形杆菌门(13.01%)、放线菌门(2.75%)。其中,DMUA 组与HUA 组相比,厚壁菌门比例显著升高,而拟杆菌门比例显著下降。
在属水平上(图5B),丰度排名前4的物种,Control组是劳特氏菌属(13.83%)、拟杆菌属(12.42%)、普拉梭菌属(10.85%)、巨单胞菌属(7.62%),T2DM 组是拟杆菌属(27.26%)、普拉梭菌属(14.16%)、巨单胞菌属(6.72%)、劳特氏菌属(4.36%),HUA 组是拟杆菌属(37.16%)、普拉梭菌属(13.36%)、克雷伯氏菌属(8.46%)、Parabacteroides(5.59%),DMUA 组是拟杆菌属(20.62%)、巨单胞菌属(17.85%)、普拉梭菌属(10.40%)、肠杆菌科未确定菌属(9.92%)。其中,DMUA组与HUA组相比,巨单胞菌属(P=0.039 9)比例升高,而拟杆菌属(P=0.003 06)和劳特氏菌属(P=0.002 46)比例降低。
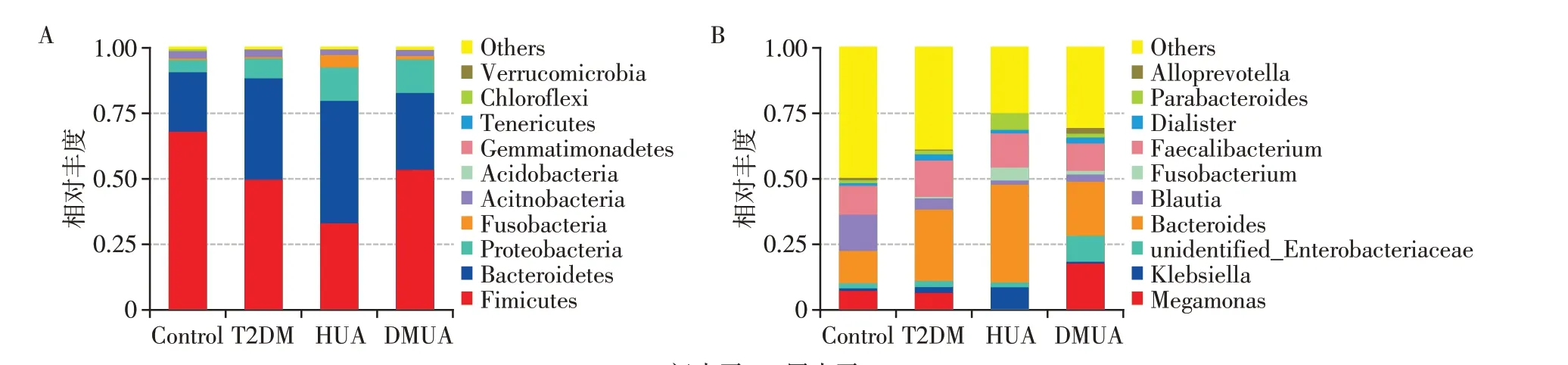
图5 门水平和属水平的相对分布情况Figure 5 Relative distribution of phylum level and genus level
2.2.6 各组间OTU差异显著性分析
DMUA 组与HUA 组相比(图6 A、B),普雷沃氏菌科(f_Prevotellaceae)和巨单胞菌属(g_Megamo⁃nas)显著增加,而拟杆菌门(p_Bacteroidetes)、拟杆菌纲(c_Bacteroidia)、拟杆菌目(o_Bacteroidales)、拟杆菌科(f_Bacteroidaceae)、拟杆菌属(g_Bacteroi⁃des)、坦纳菌科(f_tannerellaceae)和g_Parabacteroi⁃des明显减少。
DMUA 组与Control 组相比(图6 C、D),梭菌纲(c_Clostridia)、梭菌目(o_Clostridiales)、消化链球菌科(f_Peptostreptococcaceae)、g_Romboutsia 显著减少。
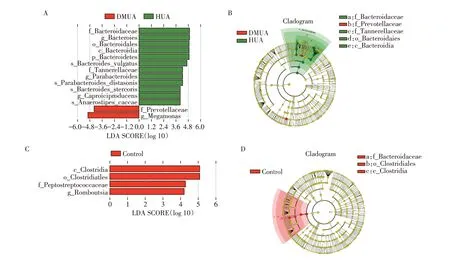
图6 DMUA组与Control组、HUA组的LDA值分布柱状图和进化分支图Figure 6 Histogram of LDA value distribution and evolutionary branching of DMUA group,Control group and HUA group
3 讨论
本研究的一般临床资料对比中显示新诊断T2DM 合并HUA 更容易合并脂代谢异常,并容易导致内脏脂肪堆积。现有多项研究均证实肠道菌群与糖尿病、高尿酸血症及脂代谢存在相关性[7-8,12-14],所以我们推断肠道菌群与新诊断T2DM 合并HUA的发生发展及其脂代谢异常可能存在一定的关系。在进一步的肠道菌群分析中,DMUA组与HUA组相比,厚壁菌门、普雷沃氏菌科和巨单胞菌属显著增加,而拟杆菌(门、纲、目、科、属)、坦纳菌科和Parabacteroides 菌属明显减少。DMUA 组与Control组相比,其梭菌纲、梭菌目、消化链球菌科、Rombout⁃sia菌属显著减少。
Shao 等[15]的有关痛风的粪便菌群及其代谢组学结果显示,痛风患者的拟杆菌属等一些条件致病菌的数量明显增加,且与尿酸排泄、嘌呤代谢及炎症反应相关的代谢产物也发生明显改变。痛风患者肠道菌群中存在的拟杆菌和梭状芽胞杆菌的变化,提示了肠道中主导菌群可能对原发性痛风的发生发展存在重要作用[16]。此外,拟杆菌门、变形杆菌门和梭状芽孢杆菌类还被证实与胰岛素抵抗及糖尿病相关[17]。在本研究中,DMUA 组的拟杆菌较HUA 组显著减少,同时其梭菌纲、梭菌目比Control组明显减少,因拟杆菌及梭菌与尿酸代谢及胰岛素抵抗均存在相关性,其丰度的变化有可能进一步促进新诊断T2DM合并HUA的发生与发展。
据文献报道[18-21],劳特氏菌属产生的代谢产物可能同时影响血糖及尿酸水平。在本研究中,DMUA 组与HUA 组在属水平相比较,其劳特氏菌属比例显著降低,提示劳特氏菌属丰度的减少可能是参与新诊断T2DM 合并HUA 发生发展的相关因素。
因代谢性疾病发病早期与厚壁菌门、梭菌纲和梭菌目的丰度变化存在相关性[22]。在本研究中,DMUA 组与Control 组的肠道菌群对比中,其厚壁菌门减少,梭菌纲、梭菌目显著减少,故推测DMUA组与Control 组之间存在的差异菌丰度的变化可能是导致新诊断T2DM 合并HUA 的发病因素之一。在使用LEFSe 方法的分析中未发现DMUA 组与T2DM组存在明显差异菌,这可能与两组患者的病程均较短有关,因本次试验样本量较少,后续可扩大样本量进一步分析。
临床资料对比中,新诊断T2DM 合并HUA 组比病程>5年组的低密度脂蛋白显著升高(P=0.02),因在入组标准的用药史中病史1~5年的患者单用二甲双胍及病史>5 年的患者为单用二甲双胍或二甲双胍联合基础胰岛素,考虑二甲双胍对脂代谢存在一定的影响,且同时会受到年龄及性别等混杂因素的影响,后续可进一步增加对照组及扩大样本量对相关指标进一步分析。
综上,DMUA 组所存在的肠道菌群丰度及组成的差异可能是新诊断T2DM 合并HUA 的发生及发展的重要影响因素之一。但由于肠道菌群复杂多样,影响菌群因素众多,本研究存在一定的局限性,仍需进一步扩大实验样本数量,可对影响的相关因素进一步细化,探索肠道菌群与新诊断T2DM 合并HUA之间的深层次关系,后续可对差异菌行代谢组学分析进一步了解细胞通路、能量传递等代谢途径是否产生变化,以期探索新诊断T2DM合并HUA的诊断、预防及治疗的方法。
