厚朴酚纳米结构脂质载体的制备及其体内药动学研究
2022-07-22许丽娜李晓蒙谈秀凤
许丽娜李晓蒙谈秀凤
(1.郑州澍青医学高等专科学校,河南 郑州450064; 2.上海中医药大学,上海201203)
厚朴为木兰科植物厚朴Magnolia officinalisRehd.et Wils 或凹叶厚朴Magnolia officinalisRehd.et Wils.var.bilobaRehd.et Wils 的干燥干皮、根皮及枝皮,主要有效成分是厚朴酚,具有中枢神经抑制、抗氧化、抗炎、治疗心肌损伤、抗肿瘤等活性[1-2],但在37 ℃下的溶解度仅为10.60 μg/mL[3],存在溶出受限、生物利用度低等问题。刘会珍等[4]通过酚磷脂复合物来改善厚朴酚溶出度及生物利用度,但该制剂黏性大,而且胃肠道水相、pH 值、酶等均会影响其稳定性[5],药物溶出度、生物利用度改善程度有限;张晓千等[6]制备的厚朴酚PLGA 纳米粒包封率仅为75.36%,仍存在一定的提升空间。
纳米结构脂质载体是在固体脂质纳米粒基础上发展而来的一种新型纳米给药技术,具有稳定性好、载药量高等特点[7-9],可促进药物体外溶出,提高体内口服吸收生物利用度[10]。本实验对厚朴酚纳米结构脂质载体处方进行优化,观察其形态,测定粒径、Zeta 电位、包封率、载药量、体外溶出情况,并研究其体内药动学,期相关制剂学研究提供新策略。
1 材料
安捷伦1260 型高效液相色谱仪(配置DAD 检测器,美国安捷伦公司);MS205DU/A 型电子天平(瑞士Mettler-Toledo 公司);HCJ-4E 型恒温水浴磁力搅拌器(常州郎越仪器有限公司);TL-150Y 型超声波细胞粉碎机(江苏天翔仪器有限公司);Master-sizer 型粒度分析仪(英国马尔文仪器有限公司);DCY-12G 型干式氮气吹扫仪(金昌实验仪器公司);TG16-G 型台式高速离心机(常州市亿能实验仪器厂)。
厚朴酚对照品(批号110729-201807,纯度98.7%,中国食品药品检定研究院);厚朴酚原料药(批号191228,纯度98%,南京泽朗医药科技有限公司)。单硬脂酸甘油酯(批号181012,国药集团化学试剂有限公司);超滤离心管(截留分子量12 000~14 000 Da,美国Millipore 公司);辛癸酸甘油酯(批号20190415,上海泰坦科技股份有限公司);大豆磷脂(批号20180914PC,上海太伟药业有限公司);泊洛沙姆 188(批 号156544891,德国BSF 公司)。SD 大鼠购自河南省动物实验中心,体质量(280±20)g,动物生产许可证号SCXK(豫)2016-0001。
2 方法与结果
2.1 厚朴酚含量测定
2.1.1 色谱条件 Diamonsil C18色谱柱(5 μm,250 mm×4.6 mm);流动相甲醇-水(60∶40);体积流量1.0 mL/min;柱温30 ℃;检测波长294 nm。
2.1.2 线性关系考察 称取厚朴酚对照品10.0 mg,转移至25 mL 量瓶中,甲醇超声溶解,放置至室温后甲醇定容至刻度,得到400.0 μg/mL 贮备液,流动相依次稀释至50.0、10.0、5.0、1.0、0.5、0.05 μg/mL,在“2.1.1”项色谱条件下进样测定。以厚朴酚峰面积(Y)对质量浓度(X)进行回归,得方程为Y=21.687 9X +0.987 2(r=0.999 9),在0.05~50 μg/mL 范围内线性关系良好。
2.1.3 供试品溶液制备 取纳米结构脂质载体混悬液1 mL 至50 mL 量瓶中,甲醇超声破乳30 s,流动相定容至刻度,0.45 μm 微孔滤膜过滤,即得。
2.1.4 方法学考察 取同一批纳米结构脂质载体混悬液,按“2.1.3”项下方法平行制备6 份供试品溶液,在“2.1.1”项色谱条件下进样测定,测得厚朴酚峰面积RSD 为1.46%,表明该方法重复性良好。取同一份供试品溶液,在“2.1.1”项色谱条件下进样测定6次,测得厚朴酚峰面积RSD为0.29%,表明仪器精密度良好。取同一份供试品溶液,于0、3、6、12、18、24 h 在 “2.1.1”项色谱条件下进样测定,测得厚朴酚峰面积RSD为0.52%,表明溶液在24 h 稳定性良好。取1 mL厚朴酚含量已知的纳米结构脂质载体混悬液至50 mL量瓶中,平行9份,加入“2.1.2”项下对照品溶液1.0、1.25、1.5 mL 各3份,甲醇超声破乳30 s,流动相定容至刻度,0.45 μm 微孔滤膜过滤,在“2.1.1”项色谱条件下进样测定,测得厚朴酚平均加样回收率分别为99.32%、100.15%、100.27%,RSD 分别为1.19%、1.42%、0.75%。
2.2 纳米结构脂质载体制备 采用熔融-超声乳化法。取50 mg 厚朴酚至圆底烧瓶中,加入处方量固态、液态脂质,75 ℃水浴磁力搅拌至药物完全溶解,作为油相;取处方量泊洛沙姆188、大豆磷脂至100 mL 蒸馏水中,75 ℃水浴磁力搅拌溶解,作为水相,在搅拌条件下将油相滴加至水相中,滴完后再敞口搅拌3 h 以除尽有机溶剂,蒸馏水补足体积至100 mL,将圆底烧瓶固定于铁架台上,置于超声仪中,在一定功率下超声处理10 min(每工作3 s 间隔1 s),迅速置于-15 ℃冰箱中10 min,取出,过0.45 μm 微孔滤膜,即得。
2.3 包封率、载药量的测定 取1 mL 纳米结构脂质载体混悬液,按“2.1.3”项下方法制备供试品溶液,在“2.1.1”项色谱条件下进样测定,计算总含量(m总)。取1 mL 纳米结构脂质载体混悬液至超滤管中(截留分子量12 000~14 000 Da),-4 ℃、12 000 r/min 离心25 min[11],取续滤液,在“2.1.1”项色谱条件下进样测定,测定厚朴酚游离量(m游离)。参考文献[8]报道,计算包封率、载药量。
2.4 粒径、Zeta 电位测定 取0.1 mL 纳米结构脂质载体混悬液,加蒸馏水至4 mL,混匀后置于比色皿中,在粒度分析仪上测定粒径、Zeta 电位。
2.5 处方筛选
2.5.1 固态脂质种类 固定液态脂质为辛癸酸甘油酯,固液脂质比为4∶1,药脂比为1∶18,泊洛沙姆188 与大豆磷脂比例为1∶1,表面活性剂用量为1%,超声功率为300 W,超声时间为20 min,分别以硬脂酸、单硬脂酸甘油酯、硬脂酸+单硬脂酸甘油酯(1∶1)制备纳米结构脂质载体,测定包封率、载药量、粒径、Zeta 电位,结果见表1。由此可知,硬脂酸作为固态脂质时平均粒径较小,但包封率、载药量、Zeta 电位绝对值较低;硬脂酸+单硬脂酸甘油酯(1∶1)制备时上述指标均不如单硬脂酸甘油酯。最终,选择单硬脂酸甘油酯作为固态脂质。
2.5.2 液态脂质种类 固定固态脂质为单硬脂酸甘油酯,固液脂质比为4∶1,药脂比为1∶18,泊洛沙姆188 与大豆磷脂比例为1∶1,表面活性剂用量为1%,超声功率为300 W,超声时间为20 min,分别以大豆油、辛癸酸甘油酯、大豆油+辛癸酸甘油酯(1∶1)制备纳米结构脂质载体,测定包封率、载药量、粒径、Zeta 电位,结果见表2。由此可知,大豆油作为液态脂质时包封率、载药量、Zeta 电位绝对值较低,粒径较大;大豆油+辛癸酸甘油酯(1∶1)制备时上述指标均不如单用辛癸酸甘油酯。最终,选择辛癸酸甘油酯作为液态脂质。

表2 液态脂质种类对包封率、载药量、粒径、Zeta 电位的影响(n=3)Tab.2 Effects of liquid lipid type on encapsulation efficiency,drug loading,particle size and Zeta potential(n=3)
2.5.3 固液脂质比 固定药脂比为1∶18,泊洛沙姆188 与大豆磷脂比例为1∶1,表面活性剂用量为1%,超声功率为300 W,超声时间为20 min,分别以固液脂质比3∶1、3.5∶1、4∶1、4.5∶1、5∶1 制备纳米结构脂质载体,测定包封率、载药量、粒径、Zeta 电位,结果见表3。由此可知,当两者比例为4∶1 时包封率、载药量最大,粒径较小,Zeta 电位绝对值大于30 mV。最终,选择4∶1作为固液脂质比。
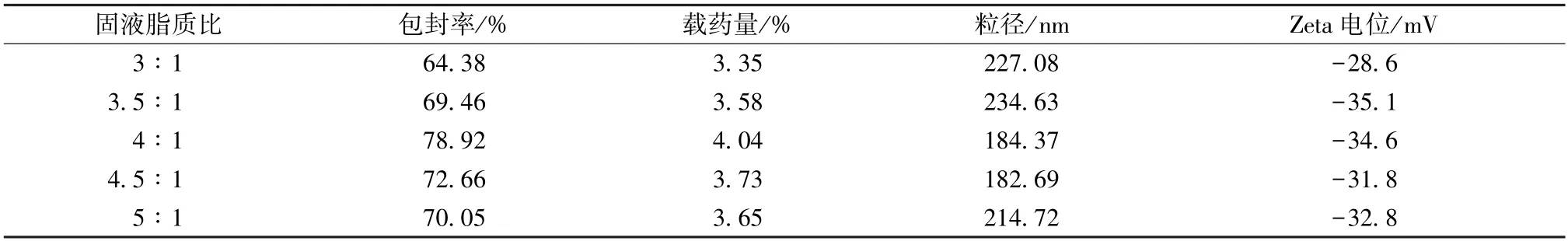
表3 固液脂质比对包封率、载药量、粒径、Zeta 电位的影响(n=3)Tab.3 Effects of solid-liquid lipid ratio on encapsulation efficiency,drug loading,particle size and Zeta potential(n=3)
2.5.4 药脂比 固定泊洛沙姆188 与大豆磷脂比例为1∶1,表面活性剂用量为1%,固液脂质比为4∶1,超声功率为300 W,超声时间为20 min,分别以药脂比1∶12、1∶15、1∶18、1∶20、1∶22制备纳米结构脂质载体,测定包封率、载药量、粒径、Zeta 电位,结果见表4。由此可知,两者比例为1∶20 时包封率、载药量、Zeta 电位绝对值相对最大,粒径较小。最终,选择1∶20 作为药脂比。
2.5.5 泊洛沙姆188 与大豆磷脂比例 固定药脂比为1∶20,固液脂质比为4∶1,表面活性剂用量为1%,超声功率为300 W,超声时间为20 min,分别以泊洛沙姆188 与大豆磷脂比例1.5∶1、0.5∶1、1∶1、1∶0.5、1∶1.5 制备纳米结构脂质载体,测定包封率、载药量、粒径、Zeta 电位,结果见表5。由此可知,改变泊洛沙姆188 与大豆磷脂比例时粒径变化较大,而载药量、包封率、Zeta 电位绝对值无明显变化规律,程度也不大。最终,选择1∶0.5 作为泊洛沙姆188 与大豆磷脂比例,此时粒径最小,Zeta 电位绝对值最大,载药量、包封率理想。
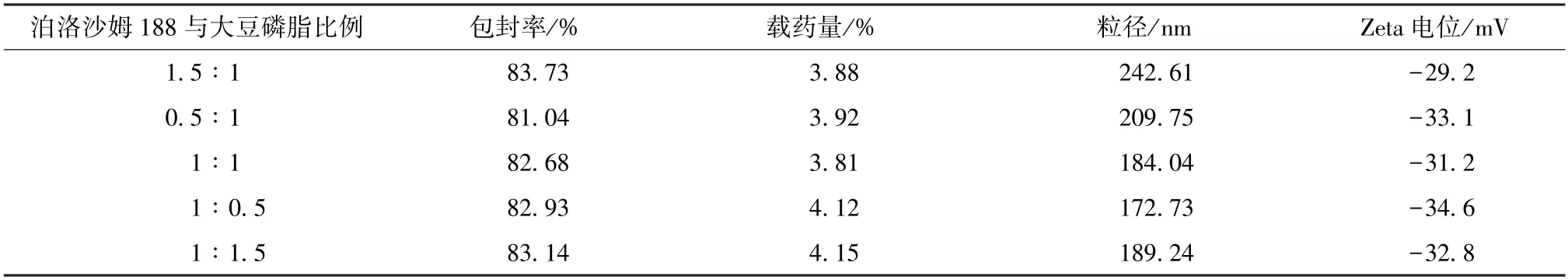
表5 泊洛沙姆188 与大豆磷脂比例对包封率、载药量、粒径、Zeta 电位的影响(n=3)Tab.5 Effects of Poloxamer 188-phospholipid ratio on encapsulation efficiency,drug loading,particle size and Zeta potential(n=3)
2.5.6 表面活性剂用量 固定药脂比为1∶20,固液脂质比为4∶1,泊洛沙姆188 与大豆磷脂比例为1∶0.5,超声功率为300 W,超声时间为20 min,分别以表面活性剂用量0.5%、0.8%、1.0%、1.2%、1.5%制备纳米结构脂质载体,测定包封率、载药量、粒径、Zeta 电位,结果见表6。由此可知,随着表面活性剂用量增加,包封率、载药量均呈先升后降的趋势,而粒径恰好相反;当其用量为1.0%时,包封率、载药量、Zeta 电位最大,粒径较小。最终,选择1.0%作为表面活性剂用量。
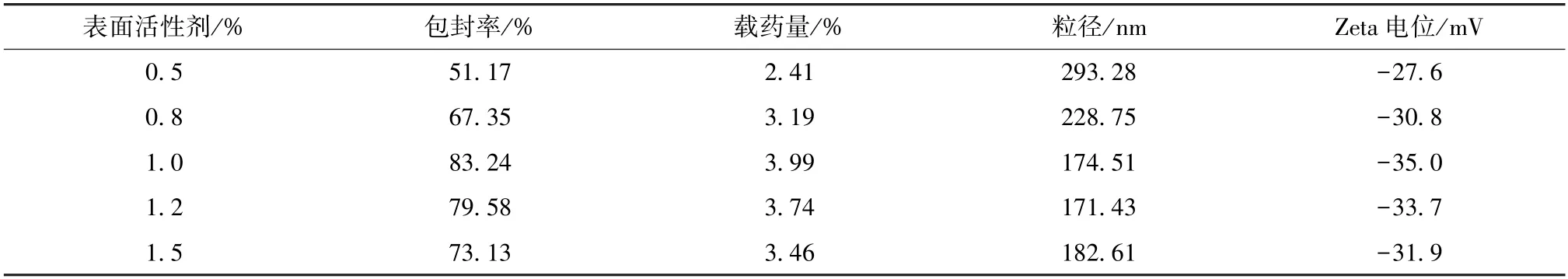
表6 表面活性剂用量对包封率、载药量、粒径、Zeta 电位的影响(n=3)Tab.6 Effects of surfactant consumption on encapsulation efficiency,drug loading,particle size and Zeta potential(n=3)
2.5.7 超声功率 固定药脂比为1∶20,固液脂质比为4∶1,泊洛沙姆188 与大豆磷脂比例为1∶0.5,表面活性剂用量为1.0%,超声时间为20 min,分别以超声功率250、300、350、400 W制备纳米结构脂质载体,测定包封率、载药量、粒径、Zeta 电位,结果见表7。由此可知,超声功率较低时粒径较大,而较高时会影响包封率、载药量、Zeta 电位绝对值。最终,选择350 W 作为超声功率。

表7 超声功率对包封率、载药量、粒径、Zeta 电位的影响(n=3)Tab.7 Effects of ultrasonic power on encapsulation efficiency,drug loading,particle size and Zeta potential(n=3)
2.6 验证试验 根据“2.5”项下结果,确定最优处方为取50 mg 厚朴酚至圆底烧瓶中,按照药脂比1∶20、固液脂质比4∶1 加入单硬脂酸甘油酯、大豆油,再加入50 mL 无水乙醇,75 ℃水浴磁力搅拌溶解,作为有机相;以泊洛沙姆188 与大豆磷脂比例1∶0.5,表面活性剂用量1.0%制备100 mL溶液,75 ℃水浴磁力搅拌溶解,作为水相,边搅拌边将有机相滴加至水相中,滴完后再敞口搅拌3 h,将圆底烧瓶固定于铁架台上,置于超声仪中,在功率350 W 下超声处理20 min(每工作3 s 间隔1 s),迅速置于-15 ℃冰箱中15 min,取出,过0.45 μm 微孔滤膜,蒸馏水补加体积至100 mL。再按照上述优化处方进行3 批验证试验,测得纳米结构脂质载体的平均包封率、载药量分别为(84.09±1.13)%、(3.92±0.17)%。
2.7 处方表征
2.7.1 透射电镜(TEM)取纳米结构脂质载体混悬液0.5 mL,加入50 mL 蒸馏水后滴加于铜网上,室温晾干,2%磷钨酸染色5 min 后观察形态,结果见图1。由此可知,纳米结构脂质载体球形,粒子之间无粘连现象。另外,TEM 观察到的是干燥纳米粒的粒径,而激光粒度仪测得的是水化粒径,故前者小于后者。
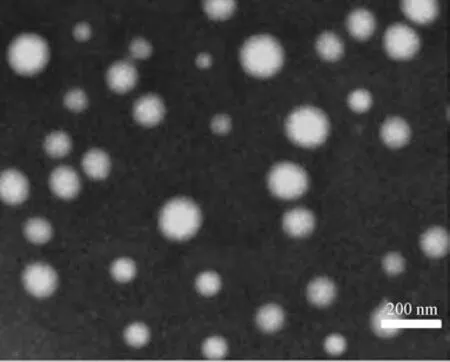
图1 纳米结构脂质载体TEM图Fig.1 TEM image for nanostructured lipid carriers
2.7.2 粒径、Zeta 电位 取纳米结构脂质载体混悬液0.5 mL,加入50 mL 蒸馏水,测定粒径、Zeta电位,结果见图2~3。由此可知,平均Zeta 电位为(-34.2±0.8)mV,粒径为(177.59±5.18)nm,PDI 为0.062±0.006。
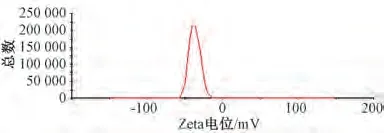
图2 纳米结构脂质载体Zeta 电位Fig.2 Zeta potential of nanostructured lipid carriers

图3 纳米结构脂质载体粒径分布Fig.3 Particle size distribution of nanostructured lipid carriers
2.8 冻干粉制备 取纳米结构脂质载体混悬液适量,加入5% 乳糖混匀,分装于5 mL 西林瓶中,在-60 ℃下预冻2 d,封口膜封口后戳6 个小孔,置于-30 ℃冷冻干燥仪中,抽真空后保存3 d,即得。取3 批冻干粉,蒸馏水复溶,测得平均包封率为(79.26±1.03)%,载药量为(3.61±0.16)%,粒径为(203.56±8.70)nm,Zeta 电位为(-29.6±0.6)mV。
2.9 体外释药研究 采用透析袋法,测得厚朴酚在0.5% 十二烷基硫酸钠溶液中的溶解度为36.64 μg/mL,达到原料药漏槽条件,故释放介质选择1 000 mL 该溶液,并设定搅拌桨转速为75 r/min,温度为(37±1)℃。取冻干粉(厚朴酚含量为5 mg)适量,加入3 mL 空白介质,转移至透析袋中,两端扎紧;取相同量厚朴酚,加入3 mL 空白溶出介质,同法操作,于0、0.25、0.5、1、2、2.5、3、4、6、8、12、18、24、30、36 h 各取样3 mL,同时注入3 mL 空白介质,0.45 μm 微孔滤膜过滤,取续滤液,测定累积溶出度,结果见图4。由此可知,原料药36 h 内累积溶出度仅为38.96%,可能是其本身颗粒较大,疏水性较强所致[12];纳米结构脂质载体后在不同时间点的累积溶出度均得到显著提高,36 h 内为78.62%。

图4 药脂比对包封率、载药量、粒径、Zeta 电位的影响(n=3)Tab.4 Effects of drug-lipid ratio on encapsulation efficiency,drug loading,particle size and Zeta potential(n=3)

图4 厚朴酚体外溶出曲线(n=6)Fig.4 In vitro dissolution curves for magnolol(n=6)
2.10 体内药动学研究
2.10.1 药液制备 取厚朴酚及其纳米结构脂质载体冻干粉适量,0.5% CMC-Na 溶液配制,即得(以厚朴酚计,质量浓度为3 mg/mL)。
2.10.2 分组、给药与采血 12 只大鼠禁食12 h,自由饮水,随机分为厚朴酚组、厚朴酚纳米结构脂质载体组,每组6只,按60 mg/kg 剂量灌胃给予“2.10.1”项下药液,麻醉后于0.25、0.5、1、2、3、4、6、8、10、12 h 眼眶采血,置于肝素化离心管中,3 000 r/min 离心2 min,取上清液,保存于-15 ℃冰箱中。
2.10.3 血浆样品处理 参考文献[6]报道,取内标溶液(称取葛根素对照品20 mg,溶于100 mL甲醇中,取适量继续用甲醇稀释至200 ng/mL,即得)、解冻血浆样品各100 μL,置于离心管中,加入乙酸乙酯2 mL,涡旋提取5 min,5 000 r/min 离心5 min,分离上层有机相至空白离心管中,45 ℃氮吹仪缓慢吹干得残渣,100 μL 甲醇复溶(将剪去的盖子扣上),混匀,转移至一次性内衬管中待测。
2.10.4 血浆对照品溶液制备 取厚朴酚对照品适量,甲醇制成 1 600、800、400、200、100、20 ng/mL溶液,分别取100 μL 至离心管中,45 ℃氮吹仪吹去甲醇,于残渣中加入100 μL 空白血浆,涡旋混匀,按“2.10.3”项下方法处理,即得。
2.10.5 线性关系考察 取血浆对照品溶液适量,在“2.1.1”项色谱条件下进样测定。以厚朴酚、内标峰面积比值为纵坐标(Y),厚朴酚质量浓度为横坐标(X)进行回归,得方程为Y=0.027 9X+7.415 3(r=0.993 2),在20~1 600 ng/mL 范围内线性关系良好。
2.10.6 方法学考察 取血浆样品适量,于0、2、4、6、12、24 h 在“2.1.1”项色谱条件下进样测定,测得厚朴酚、内标峰面积比值RSD 为4.82%,表明样品在24 h 内稳定性良好。取20、400、1 600 ng/mL 血浆对照品溶液,在“2.1.1”项色谱条件下各进样测定3次,测得厚朴酚、内标峰面积比值RSD 分别为9.62、4.35%、3.84%,表明仪器精密度良好。取20、400、1 600 ng/mL 血浆对照品溶液各3份,在“2.1.1”项色谱条件下进样测定,测得平均加样回收率分别为88.14%、93.36%、90.51%,RSD 分别为4.65%、3.11%、4.07%。取20 ng/mL 血浆对照品溶液,在“2.1.1”项色谱条件下进样测定,测得定量限(S/N =10)、检测限(S/N=3)分别为5.0、1.5 ng/mL。
2.10.7 结果分析 厚朴酚血药浓度-时间曲线见图5,主要药动学参数见表8。由此可知,原料药在12 h 时血药浓度已低于20 ng/mL,但同一时间点纳米结构脂质载体仍高于50 ng/mL;与原料药比较,纳米结构脂质载体tmax、t1/2延长(P<0.01),Cmax、AUC0~t、AUC0~∞升高(P<0.01),相对生物利用度增加至4.06 倍。

图5 厚朴酚血药浓度-时间曲线(n=6)Fig.5 Drug concentration-time curves for magnolol(n=6)
表8 厚朴酚主要药动学参数(, n=6)Tab.8 Main pharmacokinetic parameters for magnolol(, n=6)
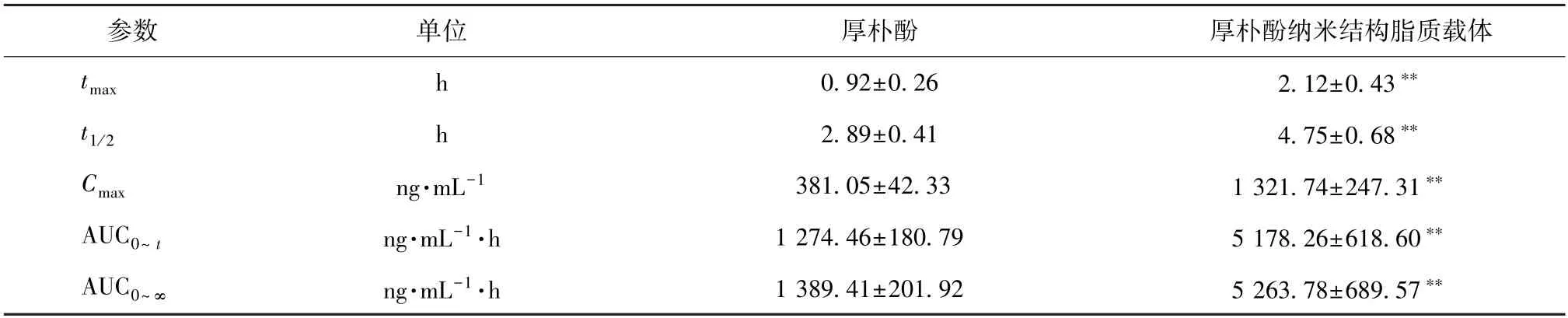
表8 厚朴酚主要药动学参数(, n=6)Tab.8 Main pharmacokinetic parameters for magnolol(, n=6)
注:与厚朴酚比较,**P<0.01。
3 讨论
前期报道,只采用固态脂质作为厚朴酚纳米载体时,包封率往往较低[5-6],故本实验联合应用固液脂质载体,包封率达(84.09±1.13)%,可能是由于它有利于形成晶体缺陷空间,可更有效地包裹药物,但液态脂质比例过高反而会影响载药量、粒径、Zeta 电位,甚至可能发生相分离[8],故最终确定固液脂质比为4∶1。包封率与脂质载体总用量呈正比,但达到一定程度后继续增加后者则会对载药量、粒径等产生不利影响,最终确定药脂比为1∶20。较高浓度的表面活性剂由于增溶作用,可使药物进入水相,从而减少被包载药物量,导致其包封率、载药量降低[8],并会影响体系的乳化效果,进而影响粒径[9],最终确定表面活性剂用量为1.0%。为确保纳米制剂的稳定性,其Zeta 电位绝对值应大于30 mV,课题组前期只采用泊洛沙姆188 作为乳化剂,所制得纳米结构脂质载体Zeta 电位绝对值在20 mV 左右,而联合应用泊洛沙姆188、大豆磷脂时其数值有所升高,可能是由于磷脂额外提供负电荷屏障,协同泊洛沙姆的空间位阻作用,有助于增加粒子之间的排斥力[13],从而改善稳定性。
大鼠灌胃给药后,纳米结构脂质载体tmax显著延长,可能是它具有较强的粘附性,易附着于胃肠道壁,从而增加滞留时间。文献[5-6]报道,厚朴酚PLGA 纳米粒、磷脂复合物、固体分散体、固体脂质纳米粒相对口服生物利用度提高至1.38~3.45倍,而本实验所制得纳米结构脂质载体可提高至4.06倍[14],可能是由于它可增加厚朴酚溶出度、体内滞留时间、药物与胃肠道接触面,从而有助于药物充分吸收[15-16],并且处方中液态脂质也有该作用[14]。
另外,采用泊洛沙姆188、磷脂作为乳化剂时,两者可附着于粒子表面。由于泊洛沙姆188 具有亲水性,磷脂同时具有亲脂性和亲水性,推测它们可能会对纳米结构脂质载体透膜吸收进入血液循环产生积极影响[16]。
