2018-2020年上海市登革1型病毒全基因组序列特征研究
2022-07-19牟嘉斌王环茹房方皓朱奕奕
王 葳,牟嘉斌,王环茹,房方皓,朱奕奕,滕 峥
登革热是由登革病毒(dengue virus,DENV)经伊蚊叮咬传播引起的急性传染病。典型的临床表现包括高热、头痛、皮疹、全身肌肉、骨骼和关节疼痛等,少数患者可发展为重症登革热,造成严重出血、休克、严重脏器损伤等后果[1]。过去50年,随着人口迅速增长、气候环境急剧变化、城市化进程的推进、全球贸易与流通加速发展等因素影响,登革热在全球范围内的发病率激增了近30倍,已在全球129个国家和地区呈地方性流行[2]。据世卫组织估计,全球每年约有3.9亿例登革热病例发生[3],将近39亿人面临着登革病毒感染风险。登革热已成为严重威胁人类健康的公共卫生问题,并造成了重大的疾病负担,2019年,登革热在全球广泛暴发,世卫组织将登革热列为全球十大健康威胁之一。
登革病毒属黄病毒科黄病毒属,基因组为单股正链RNA,全长约11 kb,两端为非编码区,内部的编码区(Coding sequence,CDS)依次编码3 种结构蛋白(C、pr M/M 和E)和7 种非结构蛋白(NS1、NS2A、NS2B、NS3、NS4A、NS4B 和NS5)[4]。根据包膜蛋白E可把DENV 分为4种血清型,分别为登革1 型 至4 型(DENV-1,DENV-2,DENV-3,DENV-4)[5],各血清型内又根据核苷酸序列差异进一步细分为多种基因型。
上海市的登革热疫情主要传染源为来自境外疫区的输入性病例,近年来受境外疫情暴发趋势影响,我市报告的输入性登革热病例数逐年增多,2017年经本中心实验室确认,报告了上海市首例本地感染病例,2018年又报告3例本地感染病例,血清型均为DENV-1型,登革热疫情防控形势正日趋严峻。本研究通过收集2018-2020年上海市哨点医院登革热疑似病例血清样本,筛选出DENV-1核酸阳性样本,进行病毒分离、全基因组扩增与测序,通过构建进化树对分离株全基因组序列进行同源性分析、核苷酸序列与氨基酸序列相似性分析、编码蛋白氨基酸位点差异分析,掌握我市输入性与本地来源病例DENV-1全基因组序列特征,为我市登革热疫情防控及相关策略制定提供科学依据。
1 材料与方法
1.1 样本来源及相关定义 样本来源为,2018-2020年上海市哨点医院采集的符合«登革热诊断»(WS216-2018)标准的登革热疑似病例血清样本。本地感染病例:发病前14 日内未离开过所在县(区),或未到过有登革热疫情报告的地区,其感染地点位于报告地区。境内输入病例:发病前14日内离开本县(区),到过本县(区)外的境内登革热流行地区的病例。境外输入病例:发病前14日内到过登革热流行的国家或地区的病例。
1.2 样本初筛与病毒分离 使用全自动核酸抽提仪及配套试剂盒(江苏硕世生物科技股份有限公司)对登革热疑似病例血清样本进行核酸抽提,采用荧光定量PCR 试剂盒(江苏硕世生物科技股份有限公司)对抽提的核酸进行登革病毒检测与分型,筛选出DENV-1核酸阳性样本。取阳性样本100μL,用细胞培养液(Gibco)按1∶10 比例稀释,接种于单层Vero-SLAM 细胞,置5% CO2培养箱37 ℃培养,每日在镜下观察细胞病变情况,收集细胞培养液上清进行核酸抽提与DENV-1鉴定,连续传代3次。
1.3 全基因组扩增与测序 针对DENV-1全基因组保守序列设计引物(表1),由生工生物工程(上海)股份有限公司合成,使用一步法RT-PCR 扩增试剂盒(invitrogen)对分离株核酸进行扩增,反应体系按说明书,反应条件为:55 ℃30 min,94 ℃2 min;94 ℃30 s,55 ℃30 s,72 ℃1.5 min,40个循环;72 ℃7 min。扩增产物由生工生物工程(上海)股份有限公司进行双向测序。全基因组核苷酸序列提交NCBI数据库,获得序列号。

表1 DENV-1全基因组扩增引物Tab.1 Primers for whole genome amplification of DENV-1

表1 (续)
1.4 进化树构建 在NCBI数据库中选取32条覆盖DENV-1 5种基因型(I-V)及代表株的全基因组序列作为参考序列(表2)。使用MEGA 7软件,选择Kimura 2-parameter模型计算核苷酸序列之间的遗传距离并进行相似性分析,进一步选择Neighbor-joining方法构建进化树,Bootstrap重复设定为1 000。

表2 DENV-1全基因组参考序列Tab.2 Reference strains of DENV-1
2 结果
2.1 病毒分离及全基因组扩增与测序 通过对2018-2020年上海市收集的登革热疑似病例样本进行初筛,共筛选出DENV-1阳性样本88份,接种Vero-SLAM 细胞后,成功获得37株分离株。对分离株核酸进行抽提,并针对DENV-1全基因组进行扩增与测序,成功得到其中31株分离株的全基因组核苷酸序列(表3)。这31株分离株的年度分布为:6株来自2018年收集的样本,24株来自2019年,1株来自2020年。病例来源为:3株本地感染病例来源,1株境内输入病例来源,输入地为广东省,27株境外输入病例来源。全长为10 734~10 736 nt,长度差异分布在3′UTR 或5′UTR,编码区长度均一致,为10 179 nt。

表3 2018—2020年上海市DENV-1分离株信息Tab.3 Information on stains isolated from dengue fever cases in Shanghai during 2018-2020
2.2 核苷酸序列进化树构建 将31株分离株全基因组核苷酸序列与32条DENV-1全基因组核苷酸参考序列构建进化树,3株分离株的基因型为G-IV型,28株分离株的基因型为G-I型(图1),3株本地感染病例来源分离株均为G-I型。

图1 2018-2020年上海市DENV-1全基因组核苷酸序列进化树Fig.1 Phylogenetic tree based on whole genomes of DENV-1 isolates
通过亲缘关系对分离株所在分支归类,3株GIV 型分离株集中在同一分支中,与2016年菲律宾参考序列(LC128301/Philippines/2016)及2001 年夏威夷参考序列(DQ672564/USA:Hawaii/2001)亲缘关系最近;28株G-I型分离株按亲缘关系远近又可进一步细分为4簇分支:包含本地感染病例来源的OM281570/038/SH/2018(以下用分离株名称038/SH/2018表示,下同)在内,共有18 株分离株集中在2015年柬埔寨参考序列(MW265679/Cambodia/2015)所在分支,占总数31 条分离株的58.06%;另外2 株本地感染病例来源的分离株042/SH/2018 与045/SH/2018,集中在2013 年 新加坡参考序列(MF033208/Singapore/2013)、2014年马来西亚参考序列(KX452058/Malaysia/2014)所在分支;2 株分离株与2014 年新加坡参考序列(KX224261/Singapore/2014)、2012年印度尼西亚参考序列(KY057370/Indonesia/2012)亲缘关系最近;6株分离株集中在2017年中国云南省参考序列(MF681692/China:YN/2017)、2016年新加坡参考序列(MF033260/Singapore/2016)所在分支。
选取编码区(CDS)、3个结构蛋白(C、pr M/M、E)与7 个非结构蛋白(NS1、NS2A、NS2B、NS3、NS4A、NS4B和NS5)对应的基因序列构建进化树,结果显示CDS、E、NS1、NS4B 基因构建的进化树中,31株分离株的基因分型及所在亲缘关系最近的分支与全基因组核苷酸序列进化树相同(图2)。

图2 DENV-1编码区、E基因、NS1基因、NS4B基因序列进化树Fig.2 Phylogenetic tree based on genes of CDS,E,NS1 and NS4B
2.3 核苷酸及氨基酸相似性分析 分析31株分离株全基因组序列与参考序列的核苷酸相似性以及编码区编码的氨基酸相似性(表4)。3株G-IV 型分离株与株G-IV 型参考序列的核苷酸(氨基酸)相似性均值为96.66%~96.86%(99.01%~99.26%),与其他基因型DENV-1参考序列的核苷酸(氨基酸)相似性均值在91.17%~91.64% (97.46%~97.61%)。

表4 核苷酸及氨基酸相似性分析Tab.4 Nucleotide and amino acid sequence identities
28株G-I型分离株与G-I型参考序列的核苷酸(氨基酸)相似性均值为96.47%~97.37%(98.78%~99.16%);与其他基因型DENV-1参考序列的核苷酸(氨基酸)相似性均值在92.67%~92.85%(97.10%~97.44%)。本地感染病例来源的038/SH/2018与MW265679/Cambodia/2015亲缘关系最近,核苷酸(氨基酸)相似性为99.44%(99.67%);另两株本地感染病例来源的分离株042/SH/2018与045/SH/2018 相互之间的核苷酸相似性为100%,与MF033208/Singapore/2013 亲缘关系最近,核苷酸(氨基酸)相似性均为99.53%(99.82%)。
2.4 氨基酸位点差异分析 以DENV-1 原型株(EU848545/USA:Hawaii/1944)作为对照株分析31株分离株各蛋白氨基酸序列差异。3个结构蛋白与7个非结构蛋白氨基酸序列均存在差异,其中NS2A 蛋白差异率最大,13.76%的氨基酸序列存在差异,其他蛋白的差异率在2.81%~10.00%。
重点分析E 蛋白氨基酸位点差异,与原型株对比,495个氨基酸残基中,31个存在差异,差异率为6.26%。我们按照全基因组核苷酸序列进化分析,结果将31株分离株分为A-E 组(表5),分别存在16、21、17、16、17个差异位点:7个变异位点(I161T、P227S、E234Q、K277T、N290D、L402F、A473T)在31株分离株均存在;此外,A 组(即G-IV株)存在的K52N、A88T、F96L、V139I、M297V、S338T、T339S、I378L、V436I 变异;N8S、T155S、S171T、V324I、V380I、I461V、M484L 在除A 组外的B-E组(即G-I株)中均存在;在此基础上,B组还存 在 K52D、T81A、H149Y、H158Y、V300M、V312L、A386T 变异;C组存在K52N、S338L、V436I变异;D 组存在K52N、A88T 变异;E 组存在K52D、T346S、E362G 变异。
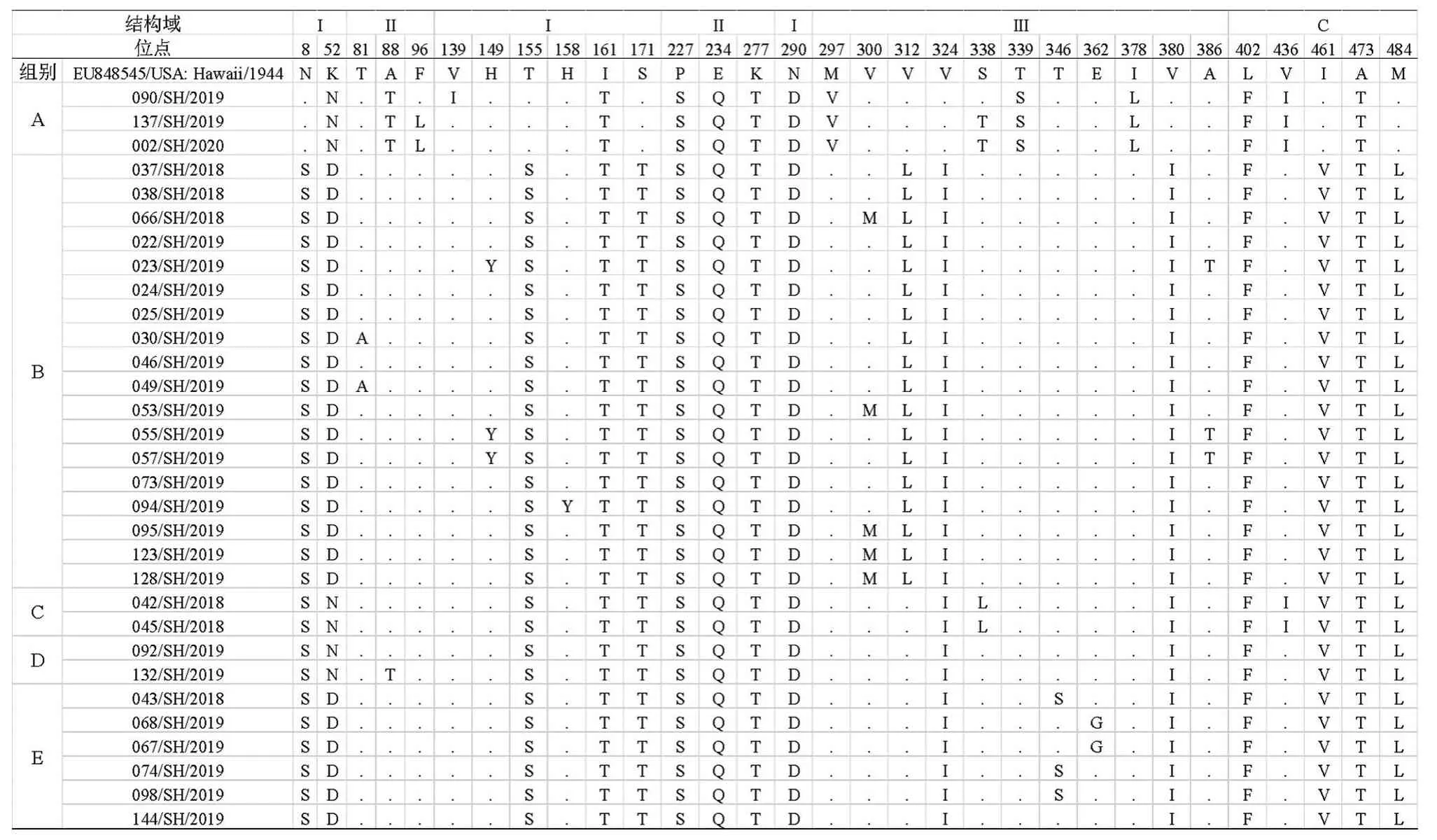
表5 E蛋白氨基酸位点差异分析Tab.5 Differences in sites of amino acid residues of the E protein between reference strain and isolates
3 讨论
DENV-1是我国目前的优势血清型,2014年广东省暴发的登革热大流行,发病人数超过4.5万人,流行株即为DENV-1[6]。上海作为全球最大的口岸城市之一,来自境外疫区的输入性病例是我市登革热疫情的主要传染源,其中DENV-1占比超过其他血清型总和。2017年与2018年经本中心实验室确认,分别报告了1例与3例上海市首例本地感染病例,血清型均为DENV-1 型。根据核苷酸序列差异,DENV-1 分为5 种基因型。G-I型主要在中国和东南亚国家流行,2014年广州与佛山地区暴发疫情中即分离到G-I型流行株[7];G-II型、G-III型分别在泰国和马来西亚等国家流行;G-IV 型主要在印度尼西亚、菲律宾等国家流行;G-V 型则主要于美洲国家[8]流行。随着近年全球登革热疫情的暴发,登革病毒基因型的地区流行特点已更显多元性与复杂性。
用于登革病毒基因分型的片段尚无标准,既往研究中,登革病毒E基因被广泛用于登革病毒的血清型与基因型分型,然而本研究中发现,E 基因进化树的拓扑结构与全基因组序列拓扑结构不完全一致,E基因进化树中的DENV-1参考序列未集中在相邻分支中(图2)。同时,依次构建其他单基因进化树,均与全基因组进化树存在差异。因此,在病毒分子分型、进化分析及选择压力等研究中,全基因组序列分析结果更加准确。
本研究分离得到的31株分离株中,3株为本地感染病例来源,于2018年8月由我市同一行政区报告。值得一提的是,2017年报告的首例本地感染病例由于样本病毒载量过低,在本次实验中未能获得有效序列信息,但流行病学资料显示,该病例也来自同一行政区。全基因组进化分析结果显示,2018年的3株本地感染病例来源分离株均为G-I型:分离株038/SH/2018 在进化树中位于与MW265679/Cambodia/2015亲缘关系最近的分支中,与邻近时间报告的境外输入病例分离株037/SH/2018高度同源,并与14株2018-2019年柬埔寨输入病例来源的分离株集中在同一分支,提示该本地感染病例与柬埔寨输入病例关联性较大;另2株本地感染病例来源的分离株042/SH/2018与045/SH/2018报告时间邻近,空间范围接近,相互之间的核苷酸相似性为100%,提示这2例本地感染病例关联性较大;同时,这2 株分离株在进化树中单独集中于MF033208/Singapore/2013所在分支,与其他输入性来源分离株亲缘关系相对较远,经NCBI数据库比对,与数据库中多株新加坡株高度同源,推测可能与新加坡输入性病例有关。综上,我们推测2018年我市报告的3例本地感染病例存在输入性病例引起本地感染可能,且存在2个不同来源。在流行病学方面,对2018-2020年人群登革病毒IgG 水平进行抽样监测,阳性率分别为2.50%、0.56%、2.54%,由于登革热感染存在自限性,推测实际感染的人数可能超过报告人数。在传播媒介方面,上海地处的气候和生态条件适宜白纹伊蚊孳生[9],2018年本地感染病例所在疫点白纹伊蚊成蚊指数在处置前为3.50~4.67只/h,布雷图指数BI为10~20[10],也为登革热本地传播提供了条件。截至目前,虽然上海市报告的本地病例较少,但病原学及流行病学研究均提示登革热存在本地传播的风险。因此,在登革热疫情防控工作中,应推进多部门联动,及时发现与处置疫情,降低输入性病例引起本地传播的风险。目前尚未在本地媒介生物样本中检测到登革病毒,一方面可能与采用的荧光定量PCR 检测方法灵敏度较低有关,另一方面,蚊虫捕获后,对分拣、保存、运输的条件要求较高,不满足相关条件,如未及时保存至液氮中,对病毒检出率有较大影响。在进一步工作中,将优化蚊虫的保存与运输流程,并探索宏基因组深度测序方法提升媒介生物样本中的病毒检出率。
28株为输入性病例来源分离株,来源呈多样性。3 株G-IV 型均为菲律宾输入,集中在与LC128301/Philippines/2016亲缘关系最近的分支中,与流行病学资料一致;其余25 株G-I型中,17株与MW265679/Cambodia/2015 亲缘关系最近,其中14 株为柬埔寨输入,1 株输入地不详,2 株(123/SH/2019、128/SH/2019)为其他地区输入,但与3株柬埔寨输入分离株(66/SH/2018、53/SH/2019、95/SH/2019)同源性为100%,分析应为同一来源;6 株与MF033260/Singapore/2016 亲缘关系最近,其中4 株为泰国输入,推测当地流行株与MF033260/Singapore/2016高度同源;2 株分别为马尔代夫与柬埔寨输入,与KX224261/Singapore/2014、KY057370/Indonesia/2012亲缘关系最近。可见,全基因组序列同源性分析结果将提供更多疫源地与流行株的信息,对病例提供的流行病学资料进行补充,以对输入性病例精准溯源,对登革热流行地提高预警等级,对途经疫区的入境人员加强健康管理,将登革热疫情防控做到关口前移。
包膜蛋白E 是登革病毒编码的重要结构蛋白,能诱导产生抗体,行使病毒与宿主细胞融合功能。E蛋白包含3个结构域(Domain):Domain I(残基1~52、133~193和281~296)组织E 蛋白结构,Domain II(残基53~132和194~280)为与细胞膜融合结构域,Domain III(残基297~394)与受体识别有关,在病毒侵入细胞和细胞嗜性中起重要作用[11]。其中,Domain I的残基132~193、280~296区域、Domain III区域对登革病毒毒力影响较大。此外,E蛋白中第67位与153位为糖基化位点,在维持病毒毒力中也起了重要作用[12]。31株分离株E蛋白氨基酸序列与DENV-1原型株(EU848545/USA:Hawaii/1944)对比,495个氨基酸残基中,31个存在差异,Domain I-III及C 端分别有9、6、11、5个差异位点,其中7个变异位点在31株分离株中均一致,推测为现流行的登革病毒在原型株基础上在进化选择压力下形成的稳定变异位点。按照全基因组核苷酸序列进化分析结果将31 株分离株分组,G-IV 株与G-I株的变异位点有较大不同,G-IV 包括16个变异位点,其中7个位点在关键毒力区域;G-I株包括26个变异位点,其中14个位点在关键毒力区域,其中T155S、S171T、V324I、V380I等4个关键毒力位点变异为G-I株特有,推测G-I株通过进化选择,尤其是关键毒力位点的变异,使得G-I株在DENV-1 中成为优势株。我们将G-I株进一步分为4组,其中占比最多的B组差异位点数也最多,包括21 个变异位点,其中H149Y、H158Y、V300M、V312L、A386T 等5个关键毒力位点变异为B组特有;S338L 为C 组特有,T346S、E362G 为E组特有。3 株本地来源感染分离株038/SH/2018、042/SH/2018 与045/SH/2018在关键毒力区域均存在7个变异位点。上述变异位点对病毒结构、毒株毒力、与宿主相互关系、致病机制等方面的影响有待进一步研究。
本研究还发现登革病毒各结构与非结构蛋白氨基酸位点均广泛存在变异,与参考序列对比,各蛋白氨基酸序列差异率在2.81%~13.76%。既往报道中,除E蛋白外,登革病毒编码的其他蛋白同样在致病过程中起重要作用。Naik等[13]发现NS4B 蛋白糖基化位点N58QN62Q 突变将显著降低病毒复制效率;Tan等[14]发现NS4A 蛋白Y41F 突变将影响病毒的适应性,减少感染性的病毒产生;Choy等[15]发现NS1蛋白G53D 突变能降低病毒的感染与传播能力。后续研究中,将对各蛋白氨基酸变异位点及相互之间的关联性做更全面的分析。
综上,本研究通过病毒分离与测序,获得31株2018-2020年上海市DENV-1全基因组序列,并对全基因组序列特征进行深入研究,为登革病毒本地传播风险评估提供病原学证据,对输入性病例精准溯源,为我市登革热疫情防控及相关策略制定提供科学依据,并为进一步的致病机制研究与疫苗研发等工作打下基础。本研究存在的不足之处为,缺乏相关病例的临床资料,不能将分离株序列特征与临床表现、疾病程度、治疗与转归等关联,希望在今后的研究中,能收集到完整的临床资料,使研究成果更具应用价值。
利益冲突:无
引用本文格式:王葳,牟嘉斌,王环茹,等.2018-2020年上海市登革1型病毒全基因组序列特征研究[J].中国人兽共患病学报,2022,38(6):507-514.DOI:10.3969/j.issn.1002-2694.2022.00.069
