Fe/α-酮戊二酸依赖型卤化酶在绿色卤化反应中的研究进展
2022-07-15王汇滨车昌丽游松
王汇滨,车昌丽,游松
(沈阳药科大学生命科学与生物制药学院,辽宁 沈阳 110016)
自100 多年前首次发现含卤素天然产物以来,目前已有5000多种含卤素天然产物被发现和鉴定,其中包括强效抗菌剂如万古霉素和氯霉素等[1]。并且大约40%的小分子药物和约30%的农用化学品含有卤素,代表性含卤素重磅炸弹药物如恩格列净、氯吡格雷和孟鲁司特等[2-4]。卤素的掺入会显著改变化合物的物理化学特性并可能影响其生物学活性、代谢途径和药物代谢动力学特征等[5]。此外碳卤键是有机合成反应中一类重要的合成砌块,可用于后续金属催化的交叉偶联反应和亲核取代反应等[6-7]。在聚合物中引入卤素单元可以有效提高其方向性和内在疏水性,含卤素材料在下一代材料设计的应用越来越广泛[8-10]。因此,小分子化合物的选择性卤化反应在药物、农药、材料等制造领域具有重要意义。然而,常见的化学卤化方法存在原子效率低、选择性低、毒性高、环境污染等问题[11-13]。
采取生物催化方法代替传统化学卤化方法会提供一种选择性更高、更高效环保的卤化物分子合成工艺路线[14-16]。生物催化卤化反应主要由卤过氧化物酶、氧气依赖型卤化酶和氟化酶等催化,根据其催化机制具体分为:遵循亲电芳香取代机制(electrophilic aromatic substitution,SEAr)的亚铁血红素依赖型、钒依赖型和黄素依赖型,遵循自由基机制(radical rebound)的Fe/αKG 依赖型和遵循亲核机制(nucleophilic substitution,SN2)的S-腺 苷-L-甲硫 氨酸[(S)-adenosyl methionine,SAM]依赖型等[17-21](图1)。在亲电反应中,卤过氧化物酶以过氧化氢和卤化物作为共底物,利用游离的次卤酸盐(XO−)作为卤化物种对芳香性和富电子底物进行非特异性卤化反应,黄素依赖型卤化酶以氧气、FADH2和卤化物作为共底物,利用固定在活性位点的次卤酸盐(XO−)作为卤化物种对芳香性和富电子底物进行区域选择性卤化反应[图1(a)]。在自由基反应中,Fe/αKG依赖型卤化酶以氧气、αKG 和卤化物作为共底物,利用卤素自由基(X·)作为卤化物种对脂肪族底物进行区域和立体选择性卤化反应[图1(b)]。在亲核反应中,SAM 依赖型卤化酶以SAM 和卤化物作为底物,利用亲核卤化物(X−)作为卤化物种对亲电子底物进行卤化反应[图1(c)]。
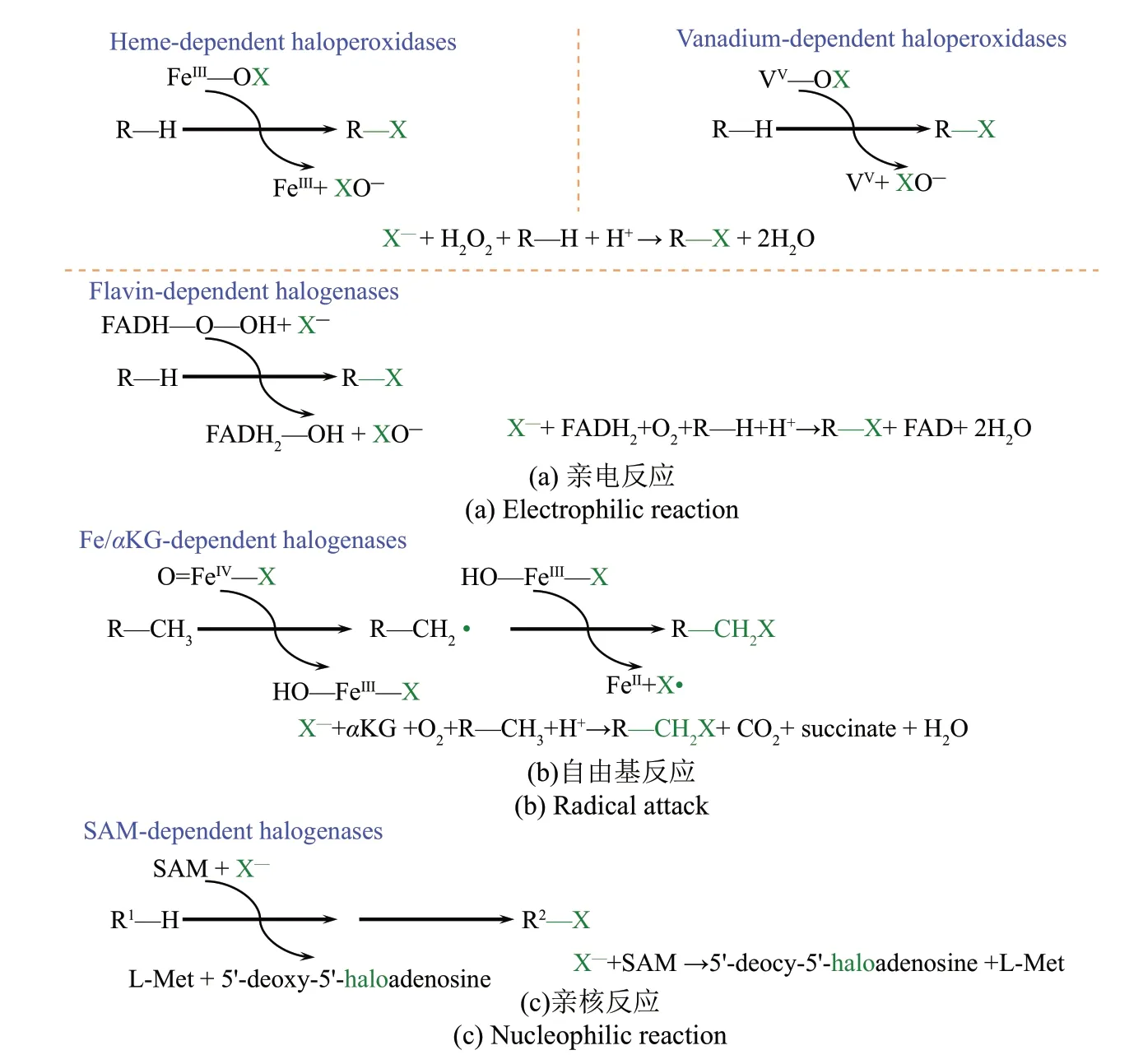
图1 不同类型卤化酶及其相关反应特征汇总Fig.1 Summary of different types of halogenases and their related reaction characteristics
本综述将重点关注基于自由基催化机制的Fe/αKG 依赖型卤化酶在绿色卤化反应中的研究进展,与其他类型卤化酶相比,它可以通过形成强氧化剂Fe(Ⅳ)=O ferrel 中间体基于自由基机制区域和立体选择性卤化脂肪族底物[22-24]。据相关统计已发现的卤化天然产物中大约一半与卤素结合的碳原子是sp3杂化的,区域和立体选择性卤化反应对天然产物高效合成至关重要,但对未经活化的C-H 键选择性卤化充满挑战[6,25-27]。卤代烷的传统化学合成方法包括利用相应的醇[28]、烯烃[29-30]和酸[31-32]进行官能团转化,现代化学合成方法包括光驱动[33-35]和过渡金属催化等[36-39]。然而,这些合成合成方法存在立体选择性低并且反应条件苛刻等问题[35,40]。相比之下,Fe/αKG 依赖型卤化酶作为生物催化剂可以在温和条件下高区域和立体选择性卤化未经活化的sp3杂化碳中心,为脂肪族底物的生物催化卤化策略开辟新的方向。本文通过对Fe/αKG 依赖型卤化酶催化的天然反应以及基于结构的蛋白质工程改造实例等展开分析总结,丰富卤化反应的酶工具箱。
1 Fe/αKG依赖型卤化酶的发现与分类
Fe/αKG 依赖型卤化酶具体分为载体蛋白依赖型(carrier protein dependent)如Barbamide 生物合成途径中的BarB1[41]、BarB2[41],Syringomycin E(丁香霉素E)生物合成途径中的SyrB2[42],Armentomycin 生 物 合 成 途 径 中 的CytC3[43],Hectochlorin(乙酰氯素)生物合成途径中的HctB[44],Coronatine(冠碱)生物合成途径中的CmaB[45],Kutzneride 2 生物合成途径中的KthP、KtzD,Curacin A 生 物 合 成 途 径 中 的CurA[46],Jamaicamide 生物合成途径中的JamE 等和独立型(free standing)如Welwitindolinone 生物合成途径中 的WelO5[47]、 WelO5*[48]、Wi-WelO15[49],Ambiguine 生 物 合 成 途 径 中 的AmbO5[50],Adechlorin 生 物 合 成 途 径 中 的AdeV[51],(−)-Acutumine 生 物 合 成 途 径 中 的SaDAH[52],βethynylserine(β-乙炔丝氨酸)生物合成途径中的BesD[53]等。基于自由基机制的Fe/αKG 依赖型卤化酶可以选择性活化含sp3杂化的碳氢键,1998 年Gerwick 课 题 组[54]在 来 源 于 巨 大 鞘 丝 藻L.majuscula的天然产物Barbamide结构中首次发现含有三卤化甲基的亮氨酸结构单元,推测其生物合成途径中可能存在未被鉴定的卤化酶催化含sp3杂化的碳氢键发生卤化反应。该课题组利用同位素标记亮氨酸并进行底物喂养实验证实亮氨酸的pro-S甲基没有与亲电试剂反应,表明碳卤键形成机制与之前鉴定的卤化酶完全不同,提出了基于自由基机制区域选择性催化Barbamide 三氯甲基部分的形成。2006 年Drennan 课题组[42]首次解析Fe/αKG依赖型卤化酶SyrB2的晶体结构,其参与来源于丁香假单胞菌Pseudomonas syringaepv.syringaeB301D 中丁香霉素E 的生物合成途径。2014 年Liu 课题组[55]在来源于Hapalosiphon welwitschiiUTEX B1830的Hapalindole类生物碱生物合成途径中发现了首例独立型Fe/αKG依赖型卤化酶WelO5。2019 年Chang 课题组[53]发现并表征氨基酸卤化酶BesD,其属于Fe/αKG 依赖型卤化酶家族。对上述载体蛋白依赖型和独立型Fe/αKG 依赖型卤化酶进行生物信息学分析,结果表明这两大分支之间同源性较低,并且不同种类的载体蛋白依赖型Fe/αKG 依赖型卤化酶同源性较高,而不同种类的独立型Fe/αKG 依赖型卤化酶同源性较低(图2)。自然界通过进化产生丰富多样的Fe/αKG 依赖型卤化酶,这类卤化酶不仅具有一定的底物宽泛性,而且有望发展成为高效的生物催化剂。
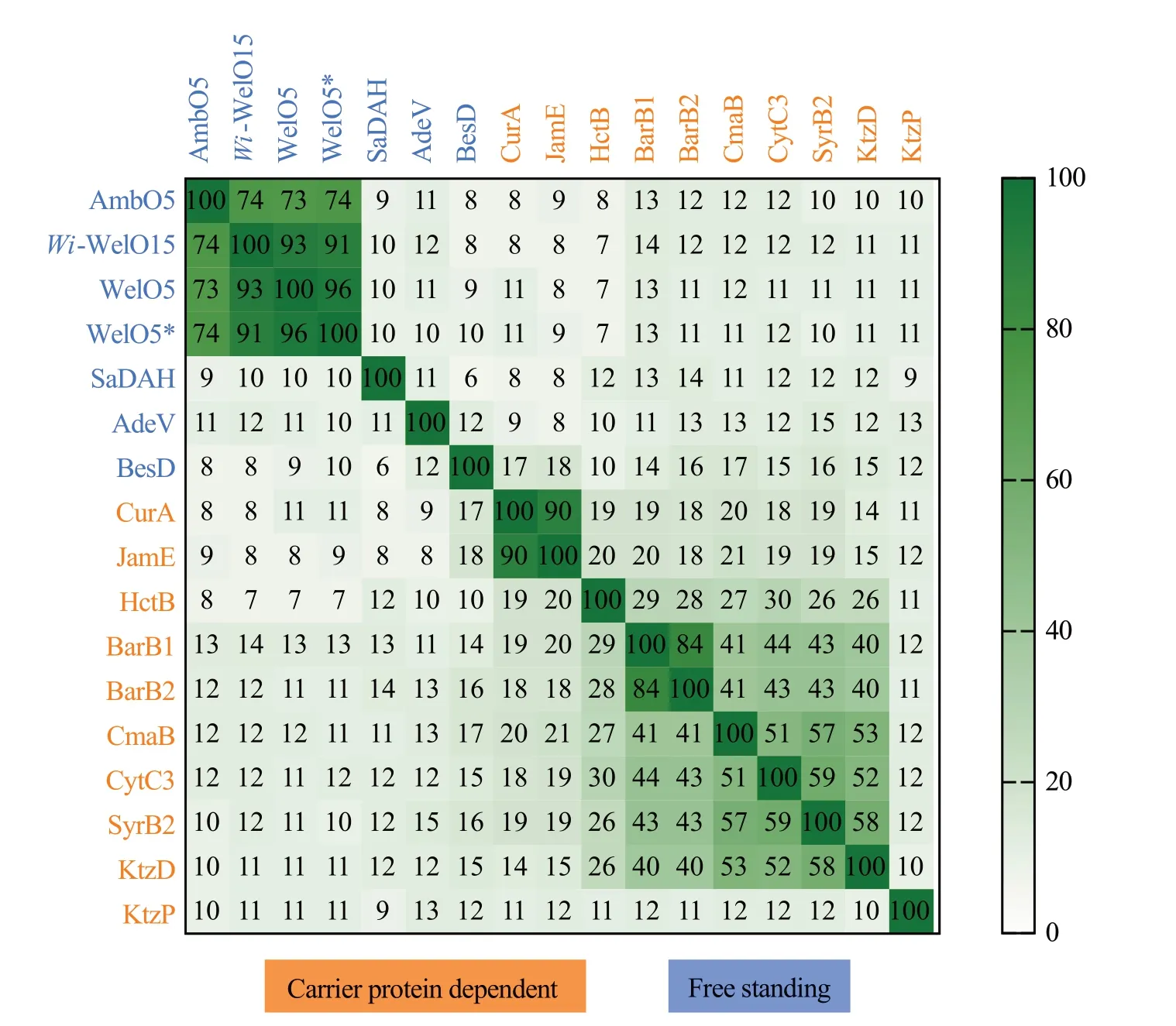
图2 Fe/αKG依赖型卤化酶序列一致性比对结果(利用MEGAX Version 10.2.4软件进行多重序列比对并通过Clustal 2.1程序分析序列一致性)Fig.2 Sequence identity matrix of Fe/αKG-dependent halogenases(Multiple sequence alignments were created by MEGAX Version 10.2.4 and the similarity percentage of the sequence identity matrix was analyzed by Clustal 2.1).
2 Fe/αKG依赖型卤化酶的天然反应
2.1 载体依赖型Fe/αKG依赖型卤化酶
2.1.1 终产物中含有卤素
载体依赖型Fe/αKG 依赖型卤化酶催化通过共价连接到磷酸泛酰巯基乙胺臂[phosphopantetheine(Ppant)arm]的酰基或肽基载体蛋白类底物。首先对非核糖体肽生物合成途径中的载体依赖型Fe/αKG 依赖型卤化酶进行归纳总结,在丁香霉素E 生物合成过程中,SyrB2 催化LThr-SyrB1 的甲基一氯化生成4-Cl-L-Thr-SyrB1 并且不接受L-苏氨酸作为底物,说明SyrB2催化底物需束缚到卤素-SyrB1 硫醇化结构域的磷酸盐臂上以进行氯化[42,56][图3(a)]。
Barbamide的生物合成途径中涉及两个Fe/αKG依赖型卤化酶BarB1 和BarB2,它们协同催化LLeu-(S)-BarA的C5位甲基发生三氯化反应。BarB2可以催化L-Leu-(S)-BarA和一氯化Leu-(S)-BarA氯化生成二氯化Leu-(S)-BarA,而BarB1可以同时催化一氯化和二氯化L-Leu-(S)-BarA 生成(2S,4S)-5,5,5-三氯-Leu-(S)-BarA,表明BarB1 和BarB2 可以催化引入不同数量的卤素原子[41][图3(b)]。
在来源于链霉菌的抗生素Armentomycin 生物合成过程中,CytC3 催化L-氨基丁酰-(S)-CytC2 的氯化反应,与SyrB2、BarB1和BarB2类似,CytC3可以催化生成γ-氯-和γ,γ-二氯氨基丁酰-(S)-CytC2,序列比对结果表明CytC3 和SyrB2 具有较高的同源性(一致性为58%,相似性为71%)[43,57][图3(c)]。
除了上述非核糖体肽合成酶相关案例外,氯化酶可以参与其他类型天然产物的生物合成途径。脂肪酰基卤化酶HctB 参与来源于巨大鞘丝藻L.majuscula中乙酰氯素的生物合成,HctB 蛋白结构包含3 个结构域,分别是N 末端Fe/αKG 依赖型卤化酶结构域、酰基辅酶A 结合结构域和酰基载体蛋白结构域。当使用与ACP 连接的己酰基作为底物时,HctB可以催化生成5-oxo-(S)-ACP、5-chloro-4-vinyl-(S)-ACP 和5,5-dichloro-hexanoyl-(S)-ACP[44][图3(d)]。在Kutzneride 2 的生物合成途径中,Fe/αKG 依赖型卤化酶KthP 区域和立体选择性催化环状底物哌嗪基的氯化反应,生成(3S,5S)-5-氯哌酸-(S)-KtzC[58][图3(e)]。
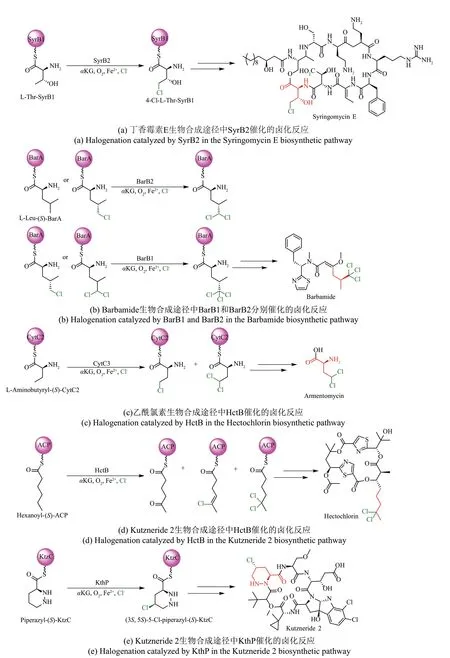
图3 载体依赖型Fe/αKG依赖型卤化酶参与含卤天然产物的生物合成途径Fig.3 Carrier-dependent Fe/αKG-dependent halogenases involved in the biosynthetic pathway of halogen-containing natural products
2.1.2 终产物中含有环丙烷结构
除上述在最终天然产物结构中含有卤素原子外,天然产物可以在其生物合成途径中通过形成隐秘的关键氯化物中间体以用于后续碳碳键形成反应,代表性案例为通过引入氯原子以形成环丙烷结构[59]。含有空间刚性环丙烷的氨基酸是一类重要的合成砌块[60],可用于激素、肽类及酶抑制剂等药物的合成过程,以避免被体内酶降解并改善生物物理特性[61],通过合成生物学途径构建非天然环丙烷氨基酸及其衍生物具有重要的理论意义和应用价值。
在丁香假单胞菌毒素冠碱生物合成途径中,Fe/αKG 依赖型卤化酶CmaB 催化L-allo-Ile-(S)-CmaD 氯化生成γ-氯-L-allo-Ile-(S)-CmaD,随后通过锌依赖性酶CmaC催化γ-消除反应生成冠状酸-SCmaD,之后冠状酸从CmaD 中释放并作为冠碱的关键生物合成砌块[45,57][图4(a)]。
Kutzneride 2 的生物合成途径与冠碱生物合成途径类似,Fe/αKG 依赖型卤化酶KtzD 催化L-Ile-(S)-KtzC 的γ 位发生氯化反应生成γ-Cl-L-Ile-(S)-KtzC,随后在黄素依赖型酰基辅酶A 脱氢酶KtzA作用下生成含环丙烷结构的(1S,2R)-allo-CMAKtzC[62][图4(b)]。
大自然在长期进化过程中产生了种类繁多的次级代谢产物,同时编码生物合成的基因簇存在平行相互作用关系,大自然采取的共同进化策略进一步丰富了天然产物的化学多样性[63-64]。来源于巨大鞘丝藻L.majuscula的Curacin A和Jamaicamide具有相同的起始生物合成途径,聚酮合酶中的卤化酶结构域CurA 和JamE 均可催化HMG-(S)-ACP 生成一氯化产物,两者序列一致性为90%,之后在脱水酶结构域CurE 或JamI 作用下生成3-methylglutaconyl-ACP。在Curacin A 生物合成途径中,上述前体在CurF结构域中脱羧酶作用下生成含α,β-烯酰硫酯结构的3-methylcrotonyl-ACP,最终在CurF 结构域中烯酰还原酶作用下发生环丙烷化。而在Jamaicamide生物合成途径中,3-methylglutaconyl-ACP 在JamJ结构域中脱羧酶作用下生成含β,γ-烯酰硫酯结构的中间体[46,65-66][图4(c)]。
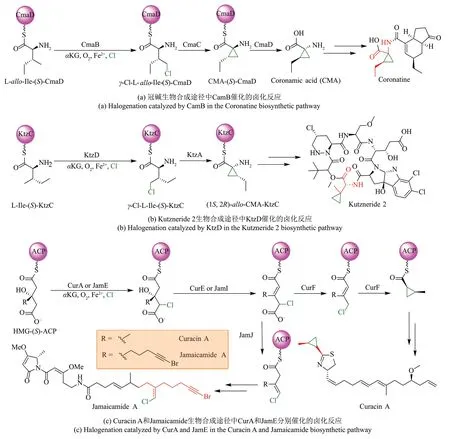
图4 载体依赖型Fe/αKG依赖型卤化酶参与催化含有环丙烷结构天然产物的生物合成途径Fig.4 Carrier-dependent Fe/αKG-dependent halogenases involved in the biosynthetic pathway of halogen-containing natural products featuring cyclopropane scaffold
2.2 独立型Fe/αKG依赖型卤化酶
来源于Hapalosiphon welwitschiiUTEX B1830的Welwitindolinone A 生物碱生物合成途径中的WelO5是第1个被鉴定的独立型Fe/αKG依赖型卤化酶,其可以区域选择性和立体选择性地氯化12-epi-Fischerindole U 和12-epi-Hapalindole C 的脂肪 碳 原子,分别生成12-epi-Fischerindole G 和12-epi-Hapalindole E,并在分子中产生一个新的立体中心[47,55]。通过基因挖掘从产生Ambiguine生物碱的Fischerella ambiguaUTEX1903 中发现了AmbO5,与WelO5相比,AmbO5具有更宽泛的底物谱,可以选择性催化Hapalindole、Fischerindole和Ambiguine等不同类型生物碱的卤化反应[50,67][图5(a)]。
独立型Fe/αKG 依赖型卤化酶不仅可以催化分子量较大的生物碱发生卤化反应,而且可以催化小分子氨基酸发生卤化反应。氨基酸卤化酶BesD参与卡特兰链霉菌Streptomyces cattleya中β-乙炔丝氨酸的生物合成,催化L-赖氨酸的γ位发生氯化反应[68]。BesD 与其他独立型卤化酶如WelO5(9%)具有较低的序列一致性,但其与Fe/αKG 依赖型加氧酶同源性更高。通过生物信息学方法以BesD 为模板进行聚类并筛选分析其同源序列,相关候选酶被分为8 个不同的簇(HalA~HalH),来自HalA~HalG的19个候选酶可以区域选择性催化不同氨基酸如L-赖氨酸、L-鸟氨酸、L-亮氨酸、L-异亮氨酸、L-正亮氨酸等的氯化反应,而来自HalH 的2种候选酶没有表现出氨基酸卤化酶活性[53],以上结果表明Fe/αKG 依赖型卤化酶可以区域选择性催化多种类型的氨基酸发生卤化反应[图5(b)]。
2019 年张勇慧课题组[51]在来源于放线菌Actinomadurasp.ATCC 39365 的含卤素天然产物Adechlorin 生物合成途径中发现了首例卤化核苷的Fe/αKG 依赖型卤化酶AdeV。AdeV 与WelO5 具有15%的相似性。AdeV催化游离核苷2′-deoxyadenosine monophosphate(2′-dAMP)发生卤化反应生成Cl-2′-dAMP。体外实验结果表明,不含磷酸基团部分的2′-deoxyadenosine(2′-dA)不与AdeV 反应,证实磷酸盐结构对于底物识别和卤化反应至关重要。此 外 两 种 磷 酸 化 核 苷2′,3′-dideoxyadenosine-5′-monophosphate 和2′-deoxyinosine-5′-monophosphate(2′-dIMP)也能与AdeV 反应并转化为相应氯化产物,但是只有天然底物2′-dAMP 活性较高,未来进一步改造Fe/αKG 依赖型卤化酶可以丰富核苷酸骨架的多样性[图5(c)]。
植物来源卤化天然产物非常罕见,2020 年Weng 课题组[52]从防己科植物中发现并鉴定Fe/αKG 依赖型卤化酶SaDAH(dechloroacutumine halogenase), 其 催 化 四 环 氯 代 生 物 碱(−)-acutumine 生物合成的最终氯化步骤。此外系统发育分析表明,DAH 在防己科植物和细菌中分别独立进化,表明不同生命领域的代谢途径采取平行进化策略,并且这类卤化反应为扩展植物化学多样性提供了新的思路[图5(d)]。
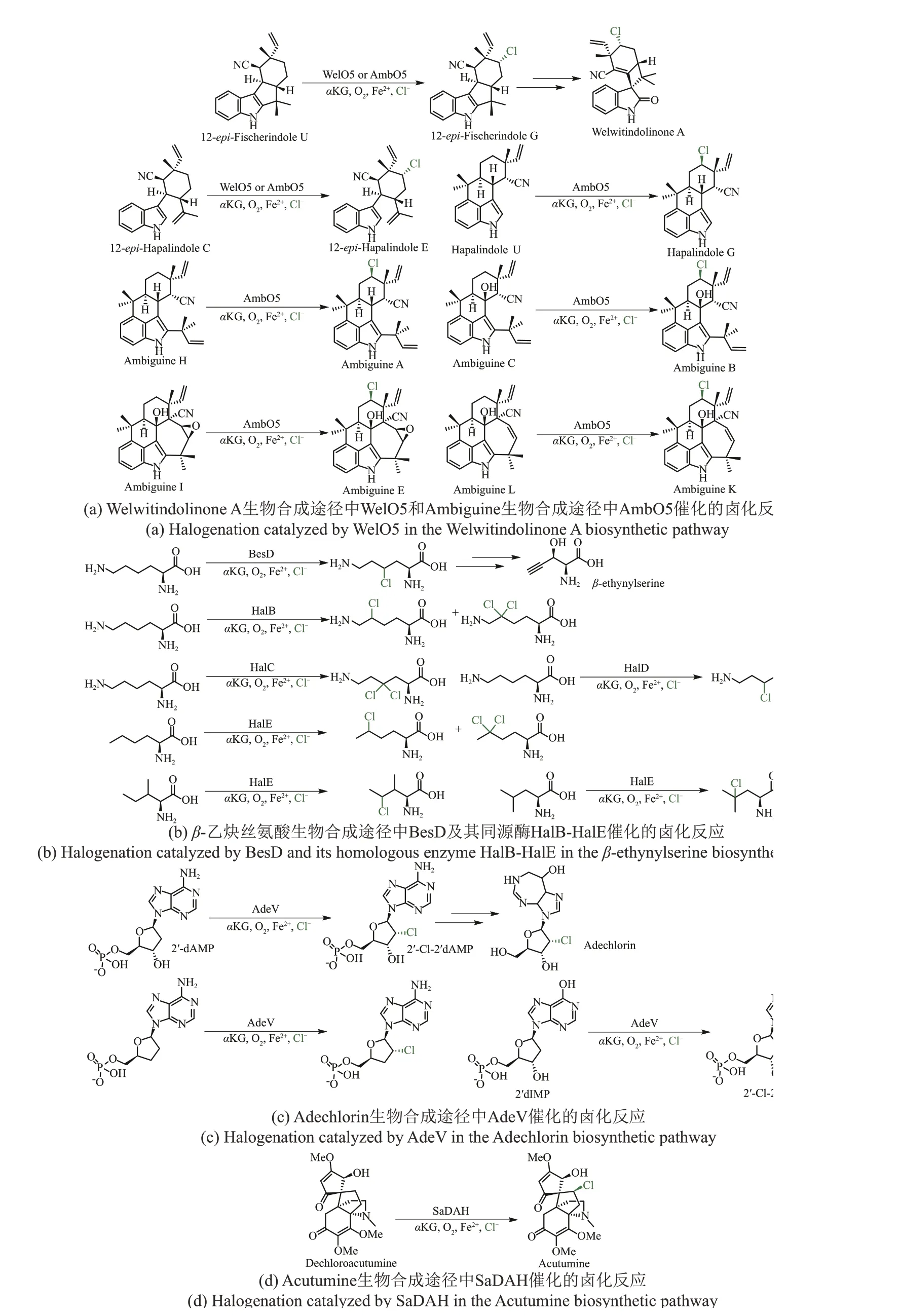
图5 独立型Fe/αKG依赖型卤化酶参与含卤天然产物的生物合成途径Fig.5 Non-carrier-dependent Fe/αKG-dependent halogenases involved in the biosynthetic pathway of halogen-containing natural products
3 Fe/αKG 依赖型卤化酶的结构与催化机理
3.1 Fe/αKG依赖型卤化酶的结构
Fe/αKG依赖型卤化酶属于Fe/αKG依赖型加氧酶超家族成员,Fe/αKG 依赖型加氧酶可以催化羟基化、环氧化、差向异构化、去甲基化、环化等多种反应[69]。迄今为止,不同类型的Fe/αKG 依赖型 卤 化 酶SyrB2[42]、 CytC3[43]、 CurA[70]和WelO5[47]、Wi-WelO15[49]、BesD[53]等的晶体结构已通过X 射线衍射进行解析,Fe/αKG 依赖型卤化酶具有Fe/αKG 依赖型加氧酶典型的DSBH(distorted double-stranded β-helix)桶状核心结构特征,底物和共底物位于DSBH 核心,外面被α 螺旋和不规则卷曲包围[69][图6(a)]。Fe/αKG 依赖型卤化酶中活性位点由铁离子、卤化物、αKG 和两个组氨酸残基组成[71],在Fe/αKG 依赖型卤化酶中铁离子与卤化物、αKG 和两个组氨酸残基形成配位作用(HXG),而在Fe/αKG 依赖型羟化酶中该卤化物位置由来自天冬氨酸或谷氨酸的羧酸盐配体取代(HXD/E)。
Fe/αKG 依赖型卤化酶在催化过程中表现出显著的构象变化。CurA 的晶体结构在开放和闭合构象的不同配体状态下被解析,在闭合构象中Cap结构域的27个残基(A40~H66)覆盖了由αKG结合引发的活性位点,之后在αKG 和氯化物作用下识别底物HMG-(S)-ACP 发生氯化作用。而在不存在αKG 情况下,CurA 处于开放构象并且Cap 结构域完全无序[70]。在独 立型Fe/αKG 依 赖 型卤化酶WelO5结构中观察到类似的构象变化,WelO5在底物结合之前采用开放构象将活性位点暴露于溶剂中,底物结合后外部α-螺旋区域(W210~Q238)移动覆盖活性位点形成闭合构象。并且WelO5晶体结构的解析丰富了对动态C末端α-螺旋区域的相关认识,该基序对于独立型Fe/αKG 依赖型卤化酶底物识别至关重要[47][图6(b)]。WelO5 中的C 末端α-螺旋区域被重组或突变,使其在结构上与AmbO5相似,构建的WelO5-AmbO5 嵌合体与野生型AmbO5 底物范围一样宽泛[50]。在底物结合方面,对于载体蛋白依赖型Fe/αKG 依赖型卤化酶,底物主要位于氯离子配体附近,而对于独立型Fe/αKG依赖型卤化酶,底物可以分别位于Fe(Ⅳ)=O中间体氧结合位点的两侧[24][图6(c)]。
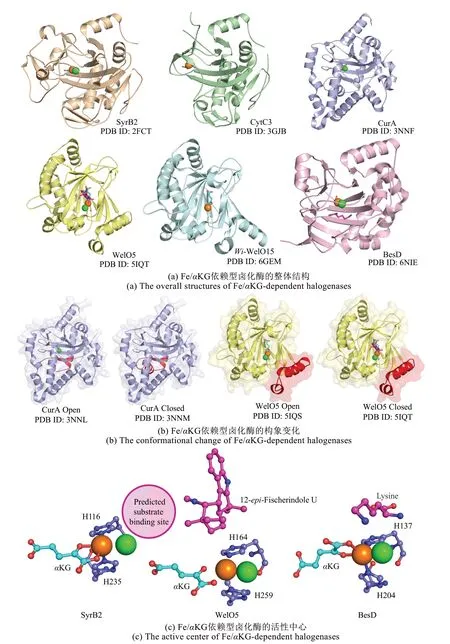
图6 Fe/αKG依赖型卤化酶的结构特征Fig.6 Structural characteristics of Fe/αKG-dependent halogenases
3.2 Fe/αKG依赖型卤化酶的催化机理
Fe/αKG 依赖型酶的非血红素亚铁离子与两个组氨酸侧链和αKG 的两个氧原子形成4 个配位作用,在羟化酶中天冬氨酸或谷氨酸的侧链羧酸部分与亚铁离子形成第5个配位作用,而在卤化酶中卤素原子代替该位置形成第5个配位作用。在没有底物时与水分子形成第6 个配位作用。Fe/αKG 依赖型卤化酶催化机制与Fe/αKG 依赖型加氧酶类似[20-21,72],通过形成高价态和短寿命的强氧化剂Fe(Ⅳ)=O ferrel中间体(Ⅵ),底物结合后触发亚铁离子中心释放水分子,随后与氧气分子结合引发催化作用,氧气分子迅速与αKG 反应形成琥珀酸盐并释放二氧化碳,从而形成高反应性Fe(Ⅳ)=O ferrel 中间体(Ⅵ)[73-75]。Fe/αKG 依赖型卤化酶基于自由基机制从底物未活化的碳氢键中攫取氢产生底物自由基和Fe(Ⅲ)-氯化物/羟基中间体(Ⅶ)。然后氯化物反弹到底物自由基生成卤化产物,而在羟化酶中羟基发生反弹生成羟化产物[76](图7)。
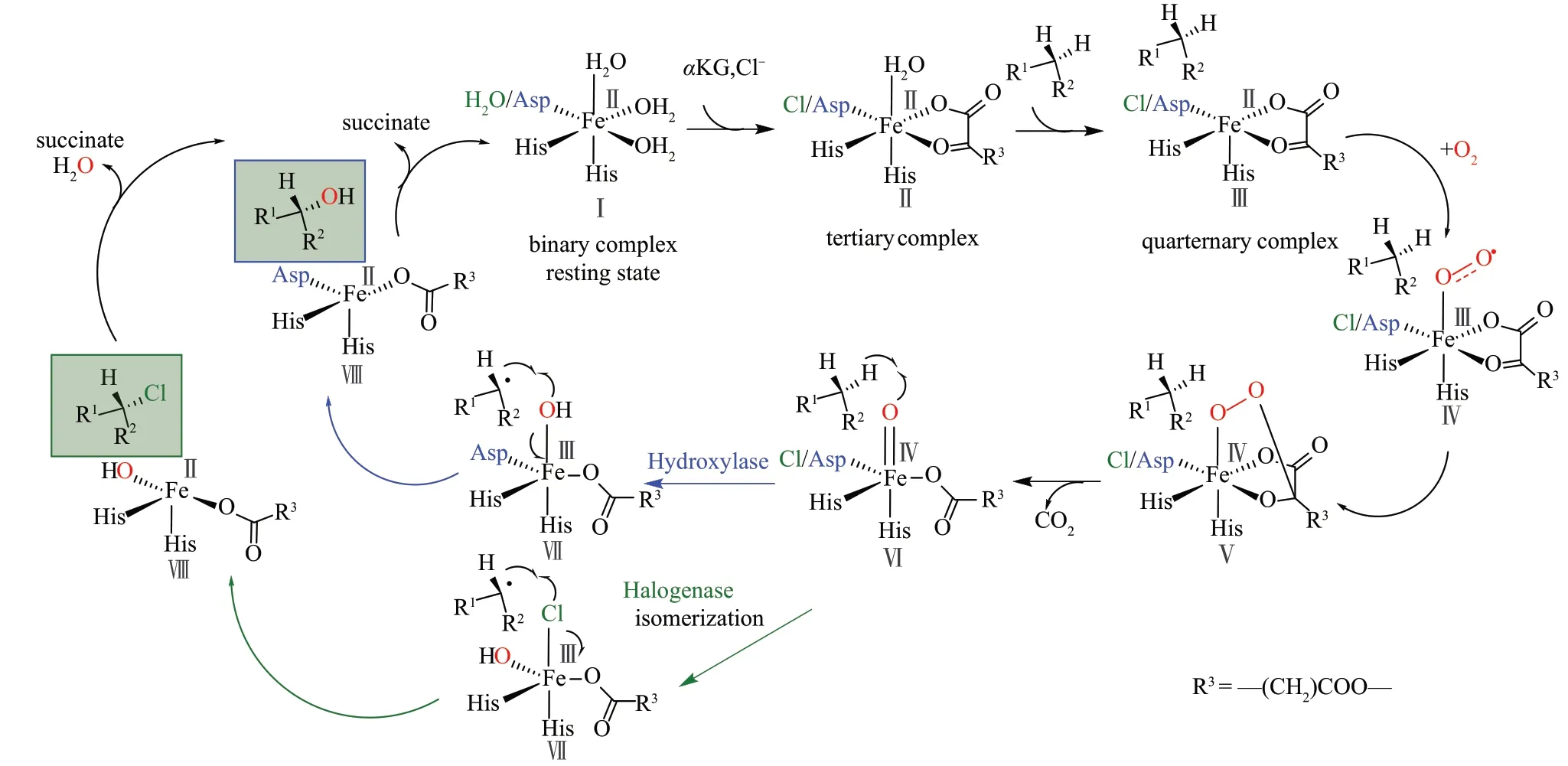
图7 Fe/αKG依赖型卤化酶和羟化酶的催化机理Fig.7 Catalytic mechanism of Fe/αKG-dependent halogenases and hydroxylases
4 Fe/αKG依赖型卤化酶的应用拓展
4.1 灵活引入卤素
卤化酶可以将不同卤素原子(氟化物、氯化物、溴化物和碘化物)引入到各类天然产物骨架中,在海洋天然产物中发现的卤化酶通常使用溴化物作为卤素来源,而在陆生天然产物中发现的卤化酶主要使用氯化物[77-79]。氟元素在自然界卤素存在范围仅次于氯元素,但氟化天然产物较为罕见,可能由于氟的电负性较大而导致自然界氟化反应较难实现[80],碘元素在自然界卤素存在丰度最低而较少存在于天然产物中。迄今为止表征的Fe/αKG依赖型卤化酶主要与天然产物的氯化反应有关,然而在含有过量NaBr的培养基中培养丁香假单胞菌时可以产生溴代丁香霉素E,此外当用更高浓度的NaBr进行检测时SyrB2表现出溴化能力,但该酶对氯化物的底物偏好性为溴化物的180 倍[81]。CytC3 可以催化其天然底物L-氨基丁酰-S-CytC2 发生溴化反应[82]。HctB在用其天然底物和高浓度KBr测定时也表现出溴化活性[44]。此外对于独立型Fe/αKG 依赖型卤化酶,WelO5也存在溴化活性可以生成(13R)-Br-12-epi-Fischerindole U[47,83]。HalB 可以催化L-赖氨酸发生溴化反应[53]。但迄今为止还没有报道过Fe/αKG依赖型卤化酶催化碘元素或者氟元素的引入(图8)。
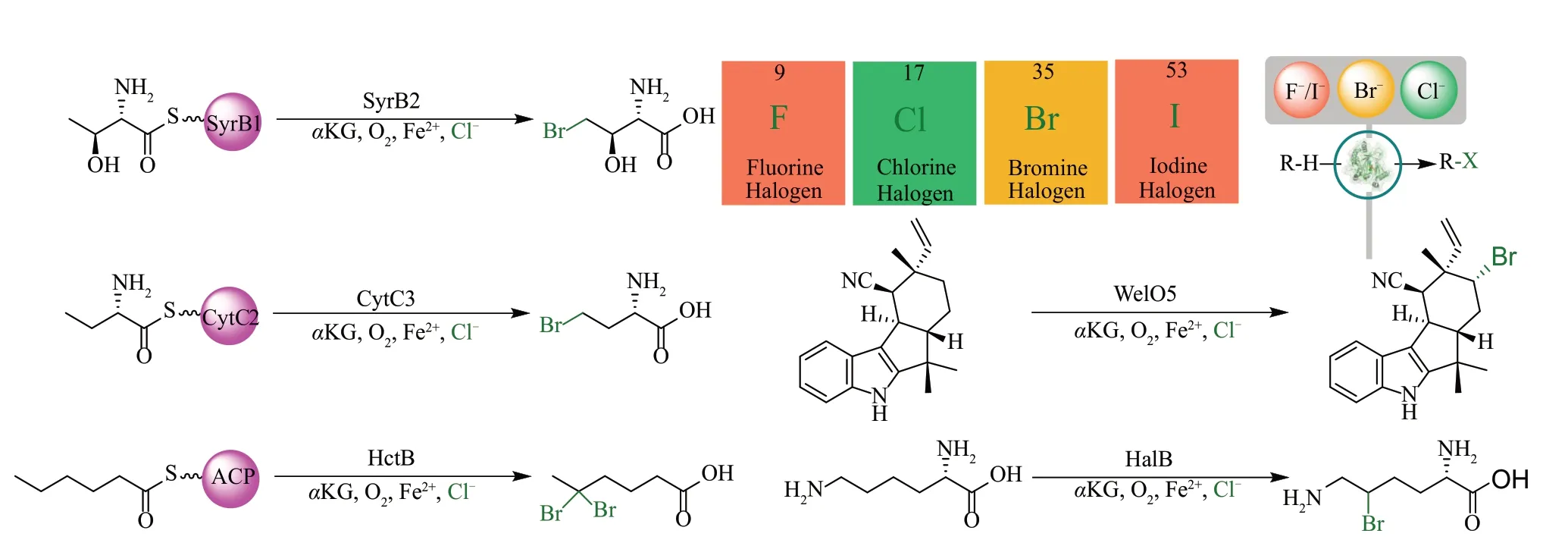
图8 Fe/αKG依赖型卤化酶催化的溴化反应Fig.8 Bromination reactions catalyzed by Fe/αKG-dependent halogenases
除了在引入氯元素和溴元素方面的灵活性外,Fe/αKG 依赖型卤化酶可以催化生物合成途径中的多种氯化反应。在Barbamide 生物合成途径中BarB2 催化L-Leu-(S)-BarA 的一氯化和二氯化反应,而BarB1催化氯化反应生成(2S,4S)-5,5,5-3-Cl-Leu-(S)-BarA,证实了这两种卤化酶在生物合成中的协同性和必要性[41]。此外,CytC3 和HctB可以催化一氯化和二氯化反应[43-44]。在未来研究中基于Fe/αKG 依赖型卤化酶引入卤素的灵活性可以进一步对其进行蛋白质工程改造以实现不同类型和数量卤素原子的引入。
4.2 拓展底物谱
大多数Fe/αKG 依赖型卤化酶与载体蛋白结合限制其作为生物催化剂的实际工业应用,然而近年发现并鉴定的WelO5 在不需要载体蛋白的情况下催化卤化反应[47,84],为Fe/αKG 依赖型卤化酶相关应用开辟了新的方向。通过对Fe/αKG 依赖型卤化酶进行蛋白质工程改造可以进一步拓宽其底物谱范围[85-86]。许多课题组已经尝试通过蛋白质工程改造WelO5 以提高生物催化活性和扩展底物范围。Boal 课题组和Liu 课题组[50]合作通过分析WelO5 的结构特征发现在催化过程中外部C 末端α-螺旋发生构象变化并向底物活性位点移动,对WelO5 和AmbO5 进行序列比对分析推测C 末端α-螺旋区域的11 个残基可能影响底物识别,由于该区域在WelO5 和AmbO5 灵活度较高,他们分别将WelO5 的N 末端与AmbO5 的C 末端结合构建WelO5-AmbO5 嵌合体,和替换C 末端α-螺旋区域的18 个残基构建WelO5-AmbO5-18 嵌合体,结果发现两种嵌合体底物范围与AmbO5 一样宽泛[图9(a)]。
鉴于WelO5 的C 末端α-螺旋区域对底物特异性的影响,Liu 课题组[48]进一步鉴定和表征来源于Hapalosiphon weltwitschiiIC-52-3 的hapalosiphon生物碱卤化酶WelO5*,该酶与WelO5 具有95%的序列一致性,仅有15 个氨基酸不同,其有11 个氨基酸位于C 末端的α-螺旋区域,结果表明WelO5*卤化反应活性比WelO5更高。
除了上述对WelO5 的C 末端区域进行蛋白质工程改造以外,Buller 课题组[87]通过分子对接确定关键位点并构建WelO5*CA2和CB2两种突变体用于在Martinelline 类似物的两个不同位点进行区域选择性卤化反应,标志着首次将WelO5 家族酶应用于非天然底物的卤化反应[图9(b)]。
在进一步扩展独立型Fe/αKG 依赖型卤化酶的底物范围研究中,Hoebenreich 课题组[49]研究发现来自Westiella intricataHT-29-1 的Wi-WelO15 能够选择性氯化非天然Hapalindoles 类生物碱。以Wi-WelO15 突变体Wi-0(V6I/D284N)晶体结构为起始结构进行4 轮定向进化,最终突变体Wi-11(N47R/V81T/A82M/A88V/V90P/S93L/S103A)和Wi-12(N47R/A82L/V90P/S93D)对12-epi-Hapalindole C底物类似物的转化率高达91%,化学选择性高达96%[图9(c)]。以上对WelO5 同源蛋白改造研究表明,独立型Fe/αKG 依赖型卤化酶的底物谱主要取决于相关关键热点残基,未来研究可以将理性设计和定向进化相结合以丰富Fe/αKG 依赖型卤化酶的底物范围。
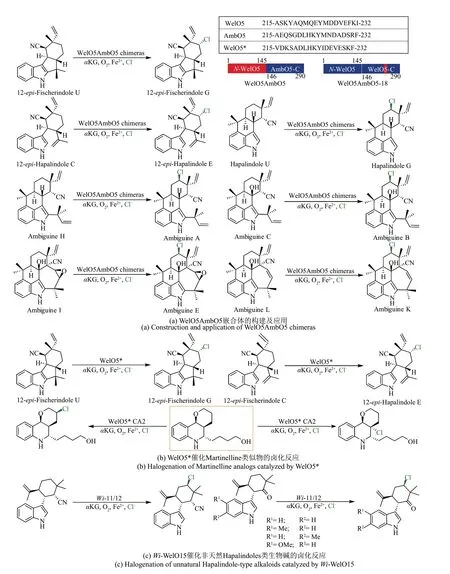
图9 独立型Fe/αKG依赖型卤化酶底物谱的拓展Fig.9 Expansion of the substrate spectrum of non-carrier-dependent Fe/αKG-dependent halogenases
4.3 Fe/αKG依赖型卤化酶与羟化酶
Fe/αKG依赖型卤化酶与羟化酶的活性位点的主要结构差异在HXG 基序。在2014 年发现WelO5 之前,Fe/αKG依赖型卤化酶仅由载体依赖型Fe/αKG依赖型卤化酶组成。因此,许多课题组尝试将Fe/αKG依赖型羟化酶功能转化为卤化酶,由于羟化酶Asp/Glu羧酸盐配体占据了卤化物相应卤素原子位置与铁离子形成配位作用,通常采取用甘氨酸或丙氨酸替换羟化酶HXD/E 基序的天冬氨酸或谷氨酸以重塑Fe/αKG 依赖型酶的二级配位球(second-coordination sphere)模拟卤化酶的HXG基序[88][图10(a)]。但是将上述策略应用于代表性Fe/αKG 依赖型羟化酶如牛磺酸双加氧酶(taurine dioxygenase,TauD)等的尝试失败了[89-90]。由此产生的突变体不仅没有发挥卤化酶的功能,而且失去其羟化酶活性,推测原因可能是TauD 突变体D101A 铁结合效率低下,或者与活性位点中的氯离子结合能力不足。
通过采取同源二级配位球检索策略,Boal课题组和Liu 课题组[91]合作利用WelO5 与其天然底物12-epi-Fischerindole U 的复合物结构以挖掘目标羟化酶,来自伯克霍尔德氏菌Burkholderia ambifaria的Fe/αKG依赖型羟化酶SadA(天然底物N-琥珀酰-L-亮氨酸)与WelO5序列一致性仅为19%,但两者活性位点高度同源。作者首先尝试用甘氨酸取代SadA 的HXD 基序中的天冬氨酸,突变体SadA D157G在NaCl或NaBr条件下卤化其天然底物天然底物N-琥珀酰-L-亮氨酸的C3位,具有高区域选择性但低卤化/羟基化比率。鉴于WelO5突变体S189A发生羟基化作用,作者尝试将SadA 中对应残基G179 突变为丝氨酸以抑制其羟基化活性,但SadA D157G/G179S 的活性比D157G 更低并且化学选择性保持不变,表明氧代中间体在野生型SadA 中与WelO5 中以不同方式稳定,羟化酶HXD 基序中的单点突变可以转换为卤化活性[图10(b)]。
此外,Buller 课题组通过对已知Fe/αKG 依赖型羟化酶进行生物信息学分析和体外实验验证,鉴定来源于细菌Sinorhizobium meliloti的脯氨酰4-羟化酶(L-prolinecis-4-hydroxylase,P4H)[92],通过引入单点突变(D108G)将其重塑为卤化酶,并且具有不同的区域选择性。通过采取4轮定向进化策略鉴定突变体SmP4H-7(V57L/S107T/D113E/T115P(off-target)/R274H),与亲本酶SmP4H-0(D108G)相比,对L-脯氨酸的氯化活性提升了98 倍。此外,突变体SmP4H-7 展现出对卤素原子引入的杂泛性,可以催化L-脯氨酸发生溴化反应,但其更偏好氯离子[93][图10(c)]。以上将羟化酶重塑为新型卤化生物催化剂丰富了碳氢键的功能化卤化策略。
上述案例表明通过利用重塑Fe/αKG 依赖型羟化酶的二级配位球策略可以成功将其转化为卤化酶。除此之外,不同课题组尝试利用上述策略将卤化酶转换为羟化酶,进一步加深对催化杂泛性的认识[88]。Drennan 课题组[42]试图通过用Asp 或Glu 替换卤化酶SyrB2 HXA 基序中的Ala 将其转变为羟化酶,然而卤化酶活性消失并且没有检测到羟基化,即使该酶保留了其结合铁的能力。除了卤化物调节和配位对空间和静电相互作用的要求外,通过计算方法对SyrB2反应性的研究表明,卤化反应时底物位于Fe(Ⅳ)=O中间体氧的远端并且靠近卤化物有利于Fe(Ⅲ)-Cl反弹发生卤化反应,而非Fe(Ⅲ)-OH 反弹发生羟基化反应[74,94-99]。这一假设通过使用非天然底物L-正缬氨酸进一步证实,L-正缬氨酸与天然底物L-苏氨酸相比含有额外的亚甲基,由于L-正缬氨酸含有较长侧链使其更接近氧代基团,因此被SyrB2羟基化[100],以上结果证实SyrB2可以氯化和羟基化不同的底物类似物,并根据特定底物定位调节化学选择性[图10(d)]。
与SyrB2 不同的是,Boal 课题组和Liu 课题组合作[47]通过用天冬氨酸或谷氨酸成功地将WelO5转化为羟化酶,突变体G166D 的晶体结构显示出在羟化酶中发现天冬氨酸与铁离子产生配位作用,此外证明了二级配位球突变体S189A 产生了羟基化和卤化产物的混合物,突出二级配位球氢键作用对化学选择性调控的重要作用[图10(e)]。
Buller课题组[87]通过以martinelline类似物为底物筛选一组独立型Fe/αKG 依赖型卤化酶(WelO5、AmbO5、 WelO5* 和 工 程 化 的SadA D157G),WelO5*主要生成羟基化产物(1%转化为氯化产物,而40%转化为羟基化产物)。为提高活性和化学选择性,该课题组通过分子对接选取WelO5* 9 个残基(N74、F77、V81、A82、I84、A88、V90、R153和I161)进行单位点饱和突变,残基A82、A88和R153的突变体文库卤化活性增加3~5倍,而残基V81和I161的突变体文库活性增加10~20倍。在筛选过程中,观察到了另一种区域选择性氯化产物。基于它们不同的区域选择性,对来自第1轮进化的最佳突变体CA1和CB1分别进行迭代饱和突变。与CA1相比,第2轮突变体CA2(V181L/I161M)在氯化活性和羟基化区域选择性方面进一步提升了10倍,而第2轮变体CB2(V181R/I161S)几乎完全催化底物氯化,并且CB2也可以催化溴化反应,但其对溴化物的偏好低于氯化物[图10(f)]。此外Hoebenreich课题组[49]发现Wi-WelO15 I84H 突变体可以催化羟基化反应,进一步证明化学选择性受二级配位球中底物定位的控制[图10(g)]。
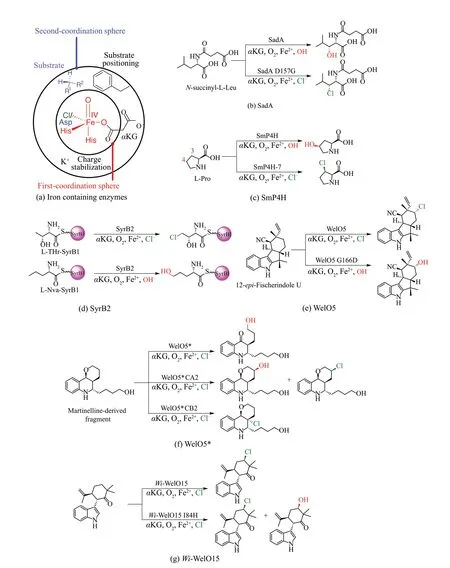
图10 Fe/αKG依赖型卤化酶和羟化酶功能互换实例(a)重塑Fe/αKG依赖型酶二级配位球的策略;(b)工程化改造羟化酶SadA催化卤化反应;(c)工程化改造羟化酶SmP4H催化卤化反应;(d)卤化酶SyrB2分别催化卤化反应和羟化反应;(e)工程化改造具有杂泛性的卤化酶WelO5*分别区域选择性催化羟化和卤化反应;(f)工程化改造卤化酶WelO5催化羟化反应;(g)工程化改造卤化酶Wi-WelO15催化羟化反应Fig.10 Function swap examples of Fe/αKG-dependent halogenases and hydroxylases(a)Strategies to reshape the second sphere region of Fe/αKG-dependent enzymes;(b)Engineering the hydroxylase SadA for halogenation;(c)Engineering the hydroxylase SmP4H for halogenation;(d)Halogenation reaction and hydroxylation catalyzed by halogenase SyrB2,respectively;(e)Engineering the promiscuous hydroxylase WelO5*for hydroxylation and halogenation,respectively;(f)Engineering the halogenase WelO5 for hydroxylation;(g)Engineering the halogenase Wi-WelO15 for hydroxylation.
类似地,Chang 课题组[53]解析BesD 与底物复合物的晶体结构时发现除了HXG 基序外,残基His134 和Asn219 分别通过与底物和氧配体的二级配位球相互作用,对化学选择性产生重要影响。在未来研究中可以通过结合理论计算结果重塑Fe/αKG 依赖型酶的二级配位球[101-102],鉴定区域选择性关键残基以实现定制化卤化反应和羟化反应。
4.4 建立新反应类型
Fe/αKG依赖型卤化酶不仅可以催化碳卤键的形成,而且能催化碳氮键的形成,例如野生型SyrB2催化脂肪族底物发生叠氮化和硝化反应[103]。SaDAH 也表现出对(−)-acutumine 的叠氮化催化活性[52]。氨基酸卤化酶HalB 可以识别叠氮阴离子生成叠氮赖氨酸[53](图11)。由HalB 和HalA 催化产生的氯代赖氨酸可以通过体外转录和翻译系统生成含9个氨基酸的短肽,体现出Fe/αKG依赖型卤化酶在利用合成生物学策略生产天然产物类似物的潜能。并且新反应类型叠氮化和硝化反应为脂肪族底物碳氢键活化形成碳氮键提供了新的生物催化途径。
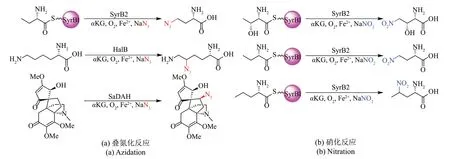
图11 Fe/αKG依赖型卤化酶的新反应类型Fig.11 Novel reaction type of Fe/αKG-dependent halogenases
5 总结与展望
不对称卤化sp3杂化碳中心具有重要研究意义,形成的碳卤键可用于有机合成后期功能化反应。自然界通过进化利用Fe/αKG 依赖型卤化酶催化脂肪族底物不对称卤化反应,在过去10 多年关于Fe/αKG依赖型卤化酶的研究中对天然产物生物合成途径进行解析不断有载体依赖型和独立型Fe/αKG 依赖型卤化酶被发现和鉴定。在功能更新方面,卤素原子的引入可以增强化合物分子的抗癌、抗真菌、抗病毒、抗炎等生物活性[104],例如抗真菌化合物丁香霉素E中氯原子的引入可以将其抗真菌活性提高4倍[105],碳卤键可以与靶标蛋白形成的卤键相互作用而影响相关活性,在药物设计中引入卤素原子从而改善相关化合物的成药性[106]。此外通过生物合成途径形成的隐秘的碳卤键可以用于构筑环丙烷合成砌块和用于合成氨基酸中末端炔基等,类似地,以碳卤键作为重要中间体将Fe/αKG 依赖型氨基酸卤化酶与后修饰酶进行级联可以合成含氮杂环、二胺、酮酸和肽类等不同类型产物[53],并且载体依赖型Fe/αKG 依赖型卤化酶也可以卤化氨基酸类底物,突出Fe/αKG 依赖型卤化酶在合成生物学多酶级联反应中的应用潜力。与其他类型的卤化酶相比,Fe/αKG 依赖型卤化酶采取独特的自由基机制区域和立体选择性卤化脂肪族底物,但目前其底物范围、催化活性等方面有待进一步提升,在未来研究中可以从以下5方面开展:
(1)新酶的挖掘与表征。WelO5 和BesD 等的发现为进一步探索新的独立型Fe/αKG 依赖型卤化酶提供重要参考,未来可以利用生物信息学方法以已知卤化酶基因作为模板并结合关键特征残基从宏基因组数据库挖掘筛选进而获得新的Fe/αKG依赖型卤化酶[107]。
(2)酶催化活性的提升。Fe/αKG依赖型卤化酶的低周转数限制其工业应用,目前主要采取在反应体系中添加抗氧化剂的策略[108]。未来研究中可以通过采取类似P450为提高周转数的定向进化方法对Fe/αKG依赖型卤化酶进行工程化改造以提升其催化活性。
(3)酶区域选择性的控制。WelO5* CA2 和CB2 两种突变体区域选择性催化martinelline 发生卤化反应[87],此外羟化酶SmP4H 可以区域选择性分别催化羟化反应和卤化反应[93]。未来可以进一步基于进化视角从酶的结构-功能角度出发鉴定调控区域选择性的关键残基以研究区域选择性机制,并有利于拓展底物范围[109]。
(4)酶反应类型的拓展。鉴于Fe/αKG 依赖型卤化酶和羟化酶两种酶家族活性位点结构高度相似,目前已有研究对羟化酶如SmP4H 进行改造使其具有羟化反应活性,对卤化酶如WelO5进行改造使其具有羟化反应活性。未来对Fe/αKG 依赖型酶的二级配位球进行重塑以实现定制化卤化反应和羟化反应[88]。此外与Fe/αKG 依赖型羟化酶类似[110],Fe/αKG 依赖型卤化酶可以催化叠氮化和硝化反应形成碳氮键[103],丰富碳氮键形成的酶工具箱。
(5)人工生物合成途径的创建。展望未来,将Fe/αKG 依赖型卤化酶引入人工生物合成途径并结合代谢工程策略可以多样性高效合成人工天然产物化合物库。此外在未来通过模拟Fe/αKG 依赖型卤化酶催化机制可以设计人工酶和仿生化学催化剂等[111-113],与其他生物合成酶进行适配创建全新的人工生物合成途径[114-116]。
综上所述,随着近年来Fe/αKG 依赖型卤化酶相关研究的不断深入,其反应类型、底物范围和选择性等不断扩展。大自然是世界上最强大的化学家,通过解析天然产物生物合成的关键酶可以丰富Fe/αKG 依赖型卤化酶类型。除此之外,未来蛋白质工程策略和机器学习结合将进一步拓展Fe/αKG 依赖型卤化酶的生物催化范围。将Fe/αKG 依赖型卤化酶发展为高效的生物催化剂并利用生物催化级联反应或化学酶法催化策略可以实现未经活化sp3杂化碳中心的选择性卤化反应,为合成生物学发展提供关键酶学催化元件(图12)。
图12 合成生物学理念指导下的Fe/αKG依赖型卤化酶工程化改造并整合至微生物细胞工厂Fig.12 Engineering of Fe/αKG-dependent halogenases and their adaption in microbial chemical factories under the guidance of the theory of synthetic biology
