Ga-Cd-O的电子结构和光学性质的第一性原理计算
2022-07-08张法碧苗国斌张荣辉
张法碧, 苗国斌, 张荣辉, 周 娟, 周 飞
(1.桂林电子科技大学 广西精密导航技术与应用重点实验室,广西 桂林 541004;2.哈尔滨工业大学 材料科学与工程学院,哈尔滨 150001)
随着科学技术的快速发展,半导体材料在光电器件、集成电路、光伏应用等领域有着广泛的应用[1-4],深受人们关注。β-Ga2O3作为新一代宽禁带半导体材料具有良好的热稳定性、化学稳定性、高击穿电压及在可见光和紫外光区的高透过率等优异性能,在纳米材料、日盲紫外探测器和高功率器件等方面具有广阔的应用前景[5-7],因此引起了科研人员的广泛关注,成为了当前的研究热点。
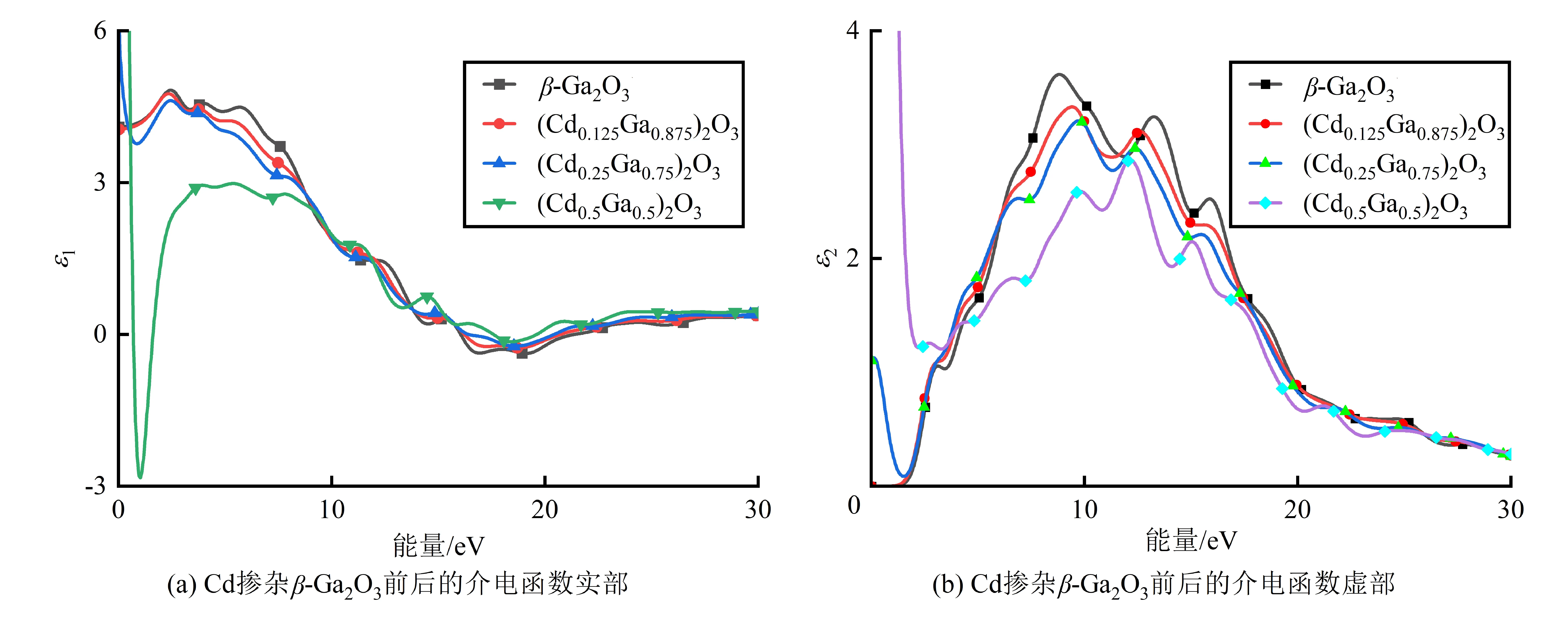
β-Ga2O3器件的能带结构与它的光电性能有密切联系,通过不同元素及不同含量的掺杂,可以灵活地设计材料体系。近年来,Ga2O3材料的研究开始从单一薄膜的制备向合金化材料转变。Bhuiyan等[8]通过金属有机化学气相沉积(MOCVD)在整个Al成分范围 (0 关于Ga掺杂CdO的相关实验目前已经有相关报道。Dakhel[10]通过蒸发法制备了低Ga掺杂CdO的薄膜样品,研究结果表明,Ga掺杂扩大了CdO的能隙。Moholkar等[11]通过喷雾热解法制备了CdO:Ga(GCO)薄膜,XRD显示薄膜具有立方结构,且随着沉积温度的升高,GCO薄膜的结晶质量逐渐提高,晶粒尺寸逐渐增大。Deokate等[12]利用化学喷雾热解技术制备了未掺杂和Ga掺杂的氧化镉薄膜,光致发光 (PL) 研究发现,纯CdO在掺杂Ga时会由紫外线发射转变为绿色发射。Li等[13]通过脉冲激光法制备了低Ga掺杂浓度的CdO薄膜,随着Ga掺杂浓度增加,光学间隙在2.49~2.85 eV变化。Liu等[14]在室温下通过射频磁控溅射在Ga的整个成分范围内合成了(CdxGa1-x)2O3合金薄膜,XRD显示具有高Ga含量(x> 0.3)的合金薄膜是非晶态;同时当Ga 含量在0.3~0.5时,(CdxGa1-x)2O3合金表现出高电子迁移率,在整个组成范围内,(CdxGa1-x)2O3合金具有 2.2~4.8 eV的带隙可调范围。 关于Cd掺杂的Ga2O3方面的相关研究较少。Yanagi等[15]通过射频 (RF) 磁控溅射制备了Cd-Ga-O薄膜,薄膜带隙具有从2.5~4.3 eV的可调范围,同时还保持着高电子迁移率。Xiao等[16]通过溶液法制备了非晶Ga2O3:CdO 薄膜,随着Cd从增加到20%,光学带隙从4.83 eV 减小到4.05 eV,且随着Cd掺杂浓度的增加,相应的 TFT 器件显示出较大的导通电压负移,同时所有 TFT 都对260 nm 深紫外光和UV/VIS 敏感。 关于Ga2O3掺杂的基于密度泛函理论(DFT)第一性原理计算已有相关报道。Minseok等[17]使用混合泛函的第一性原理计算发现,添加模拟合金系统中的掺杂剂可调节α-Ga2O3的带隙,提出了氧化镓基半导体的间接到直接带隙跃迁的新策略。Peelaers等[18]使用混合密度泛函理论研究了自由载流子对Ga2O3性质的影响。Kumar等[19]通过第一性原理计算研究了块状β-Ga2O3晶体中的热电效应。Zhang等[20]使用基于密度泛函理论的第一性原理计算了本征β-Ga2O3和N掺杂β-Ga2O3的能带结构、态密度、电子密度差和光学性质。计算结果表明,N掺杂是一种非常有前途的获得 P型β-Ga2O3的方法。Dong等[21]采用基于密度泛函理论的第一性原理计算来研究α-Ga2O3中的单元素(Si,Sn,Mg)掺杂,为α-Ga2O3的进一步应用提供了参考。孟德兰等[22]采用GGA+U的方法研究了纯β-Ga2O3和Ti掺杂β-Ga2O3的电子结构和光学性能。目前,关于Cd掺杂β-Ga2O3的相关理论研究相对较少,对于其掺杂体系的电子结构、光学性质的研究有待进一步深入研究。因此,基于密度泛函理论的第一性原理方法,计算并分析了Cd掺杂β-Ga2O3的电子结构、光学性质,为进行相关的β-Ga2O3掺杂Cd的实验提供相应的理论参考。 β-Ga2O3为单斜晶系,属于C2/m空间群[23]。计算采用的1×1×1β-Ga2O3晶胞结构模型如图1(a)所示,由8个Ga原子和12个O原子组成,其中Ga原子有2种不同位置,分别用Ga1和Ga2表示。通过用不同个数的Cd替换Ga原子,可得到Cd掺杂量不同的(CdxGa1-x)2O3合金。但在构建Cd掺杂量不同的(CdxGa1-x)2O3合金前,先分别使用一个Cd原子替换不同的Ga原子位置,即Ga1和Ga2,如图1(b)为Cd掺杂Ga2后的结构模型,然后计算1个Cd原子分别取代Ga1、Ga2后相关的(CdxGa1-x)2O3总能量,计算结果表明,Cd替代Ga1后体系的总能量(-20 901.703 eV)高于Cd替代Ga2后得到的体系总能量(-20 902.198 eV)。物质体系的总能量越低,对应的物质结构就越稳定,因此,Cd掺杂β-Ga2O3时优先替换Ga2。根据这样的思想,构建了Cd掺杂量不同的(CdxGa1-x)2O3合金。 计算均在Materials Studio软件的Castep模块中进行。采用超软赝势来描述电子与离子核之间的相互作用,采用广义梯度近似GGA下的PBE计算方案描述交换相关相互作用[24]。将2×8×4的K点网络用于在布里渊区采样,对于所有计算,平面波的截止能量Ecut设置为700 eV。对晶胞模型采用BFGS算法进行几何优化,其收敛标准为:原子的最大位移为0.001 Å,SCF容差为1.0×10-5eV/atom,原子的相互作用力低于0.03 eV/Å,晶体内应力为0.05 GPa。相同的收敛标准在Ge和F掺杂α-Ga2O3[25]及Ti 掺杂β-Ga2O3[26]的第一性原理计算研究中也被采用。本次计算分别选取O原子的 2s22p1态、Ga原子的 4s24p1态、Cd原子的4d105s2态来作为价电子进行计算。 在对相关模型进行几何优化计算后,得到了纯β-Ga2O3和Cd掺杂的β-Ga2O3的稳定结构。表1为纯β-Ga2O3和(CdxGa1-x)2O3(x= 12.5%、25%、50%)体系结构优化后的晶格常数。纯β-Ga2O3的晶格常数分别为a= 12.48 Å,b= 3.09 Å和c= 5.89 Å,与实验值[27]非常吻合,且相对误差不超过2.21%,因此,计算方法是相对准确的。从表1可看出,Cd掺杂前后β-Ga2O3和(CdxGa1-x)2O3(x= 12.5%、25%、50%)体系结构中夹角α=γ= 90°,依然是单斜结构,并未发生相变,且随着Cd掺杂量的增加,晶格常数也逐渐变大,导致体系的晶胞体积逐渐变大。这可能是因为Cd2+的离子半径(0.095 nm)略大于Ga3+的离子半径(0.062 nm),当离子半径较小的Ga3+被离子半径较大的Cd2+替代后,掺杂Cd后的(CdxGa1-x)2O3(x= 12.5%、25%、50%)体系的晶胞体积应当有所增加。类似的情况也有报道,研究人员在Mg掺杂的 Ga2O3的实验中[28]发现,由于Mg2+的离子半径(0.072 nm)大于Ga3+的离子半径(0.062 nm),Mg掺杂后Ga2O3的晶格参数有所增加。此外,在Zn掺杂Ga2O3的研究中[29]也发现了Zn掺杂后的Ga2O3的晶格参数增加,这是因为Zn2+离子半径(0.074 nm)大于Ga3+离子半径(0.062 nm),从而导致晶格间距扩大。从表1还可看出,随着Cd掺杂浓度的增加,(CdxGa1-x)2O3(x= 12.5%、25%、50%)体系结构的总能量增大,这表明掺杂体系的稳定性在逐渐降低。 图2为β-Ga2O3及(CdxGa1-x)2O3(x= 12.5%、25%、50%)体系的能带结构,其中费米能级位于纵坐标0处。图2(a)中β-Ga2O3的导带底部的位置在G点处,而价带顶则位于Z点,符合间接带隙半导体的特性,因此β-Ga2O3是间接带隙半导体,这与之前的报道相符合[30]。经计算得到的β-Ga2O3带隙宽度为1.973 eV,但它远小于β-Ga2O3的实际禁带宽4.8 eV,这是由于在用DFT理论计算晶体结构时,激发电子的交换相关势被低估了[31],因此计算出来的禁带宽度要比实际的禁带宽度小。具体到本实验,对β-Ga2O3晶体而言,主要是因为在计算过程中Ga 3 d态的能量过高,造成Ga 3 d态与O 2p态之间相互作用的增大,结果使价带带宽增大,带隙偏低[32]。虽然GGA近似的计算方法会导致带隙偏低,但这不影响对能带问题的定性分析。当Cd掺杂浓度为12.5%时,能带如图2(b)所示,禁带宽度从1.973 eV减小到1.634 eV,价带顶位置发生了变化,此时价带顶和导带底位于同一点(G),(Cd0.125Ga0.875)2O3变为直接间隙半导体;当掺杂浓度增加到25%时,能带如图2(c)所示,此时禁带宽度增加到1.734 eV,导带底和价带顶仍位于G点,即仍为直接间隙半导体;当掺杂浓度为50%时,能带如图2(d)所示,禁带宽度进一步增加到1.86 eV,此时(Cd0.5Ga0.5)2O3的价带最高点不再是G点,于是(Cd0.5Ga0.5)2O3由直接带隙半导体变成间接带隙半导体。濮春英等[33]在对Cd掺杂ZnO的电子结构的第一性原理研究中发现,随着Cd含量的提高,合金的禁带宽度逐渐减小,且合金的禁带宽度随掺杂比例变化满足抛物线方程 表1 结构优化前后模型的晶格参数、体积和体系总能量 图2 Cd掺杂β-Ga2O3前后的能带结构 Eg(x)=3.28-5.04x+4.60x2。 基于GGA+U方法,He等[34]在研究In掺杂对β-Ga2O3的结构特性的影响时发现,随着In掺杂含量的提高,β-In2xGa2(1-x)O3的晶胞体积增大,带隙减小,认为带隙减小的主要原因是掺杂体系结构变形的影响。但本文中,禁带宽度Eg随Cd掺杂浓度先减小后增大的原因尚不明确,需要进一步研究。 图3(a)为β-Ga2O3的态密度图,结合图2(a)可看出,价带分为3个区域:上价带主要由Ga 4p、4s和O 2p态形成,位于费米能级下方0~7.0 eV左右;位于-11.2~-13.5 eV 的中价带主要由 Ga 的3d态电子贡献;位于-16.2~-18.8 eV 的下价带,主要是来自O的2s态电子贡献,Ga的4s及4p轨道上的电子也有少量贡献。导带底部主要来自Ga原子的4s态。众所周知,β-Ga2O3的光电特性主要由价带顶和导带底决定,因此不讨论中价带和下价带。 图3(b)~(d)为了Cd掺杂β-Ga2O3后的态密度,结合图2(b)、(c)、(d)可知,Cd的5s、4p、4d态分别在0~-7.6 eV、-11.6~-13.2 eV、-16.0~-17.9 eV都有分布,与O的2s、2p态的态密度有所重叠。掺杂后Ga原子的原子态密度有所减小,Cd原子的原子态密度有所增加,对于(CdxGa1-x)2O3合金的态密度分布,价带顶主要由Cd 4d态和O 2p态占据,而导带底主要由Cd 5s态和Ga 4s态占据。相对于本征β-Ga2O3,掺杂后的(CdxGa1-x)2O3合金的价带顶向高能方向移动,而导带底有所降低。类似的现象在Ca掺杂α-Ga2O3也有报道,由于Ca 3p和O 2p轨道的耦合,导带底向下运动,导致了(CdxGa1-x)2O3合金的带隙减小[35]。 图3 Cd掺杂β-Ga2O3前后态密度 在线性光学响应范围内,固体宏观的光学响应一般可用光的复介电函数ε(ω)=ε1(ω)+ iε2(ω)来描述,其中ω为光的频率。介电函数实部ε1(ω)可通过Kramer-Kronig散射方程和ε2(ω)求得。图4为本征β-Ga2O3和 (CdxGa1-x)2O3(x=12.5%、25%、50%)合金体系的介电函数实部和虚部。从图 4(b)可看出,本征β-Ga2O3的介电函数虚部有2个主要的峰,分别位于8.8、13.3 eV处,对应着价带中 O 2p到导带中 Ga 4s的跃迁,在15.9 eV处还有一个较小的峰,是由Ga 3d电子激发到导带的Ga 4s轨道产生的。随着Cd含量的增加,(CdxGa1-x)2O3体系的主峰位置向高能区移动,这可能与掺入Cd后晶格发生了畸变及形成的杂质能级有关。 图4 Cd掺杂β-Ga2O3前后介电函数 本征β-Ga2O3和Cd掺杂的β-Ga2O3在能量范围内(0~50 eV)的吸收系数如图5(a)所示。在本征β-Ga2O3和(CdxGa1-x)2O3(x= 12.5%、25%、50%)体系中,都显示了在16.6 eV左右的高能量峰,这些峰值与从价带O 2p占据态到导带Ga 4s未占据态的带间跃迁有关。本征β-Ga2O3和掺Cd的β-Ga2O3的光吸收范围主要集中在2~45 eV。掺杂Cd后材料的吸收峰强度在6~25 eV范围内明显降低。 图5(b)为纯β-Ga2O3和Cd掺杂的β-Ga2O3在能量为0~50 eV范围内的反射率。而对于本征β-Ga2O3,在整个波段都有一定程度的反射,且在19 eV附近出现了一个主强峰。对于Cd掺杂β-Ga2O3体系,主峰也位于19 eV附近,但反射率低于本征β-Ga2O3。在4~30 eV范围内,反射率随Cd掺杂浓度的增加而降低。 采用基于密度泛函理论的第一性原理计算方法,研究了Cd掺杂对β-Ga2O3晶格结构、电子特性以及光学性质的影响。研究结果表明,在β-Ga2O3中掺杂Cd后,由于Cd离子半径大与Ga离子半径,导致(CdxGa1-x)2O3体系的晶格常数增大,稳定性降低。同时Cd的掺入能够减小β-Ga2O3的带隙,带隙的降低是由于产生了更多的空穴来接受更多的自由电子,表明Cd可能是β-Ga2O3潜在的P型掺杂杂质,这需要进一步的实验来证明。另外,Cd掺杂使得(CdxGa1-x)2O3体系的吸收系数和反射率在可见光区和紫外区都明显降低,从而使其透光率增加。这表明Cd掺杂β-Ga2O3在多波段的光电探测器方面具有应用潜力。1 构建模型与计算方法
2 结果与讨论



3 结束语
