纳米铝粉包覆材料选择的分子动力学模拟
2022-06-04欧阳的华张浪浪毛睿晋庞维强
欧阳的华,张浪浪,毛睿晋,庞维强
(1.西安建筑科技大学 资源工程学院,西安 710055; 2.西安近代化学研究所, 西安 710065)
1 引言
在现代军事行动中,传统的大面积武装冲突对抗减少,恐怖袭击事件呈现高发态势。在此情况下,特种弹药作为解救人质处理突发事件的常用武器装备发挥了重要作用。铝粉作为一种含能材料,因具有较高的燃烧热值和更好的燃烧性能常用作特种弹药的可燃剂,近年来被国内外研究者广泛关注。现阶段已广泛应用于推进剂、烟火药等国防工业体系中。因小尺寸效应纳米铝粉相比于微米铝粉具有更大的比表面积和反应活性,但纳米铝粉极易在空气中氧化生成氧化铝,从而导致铝粉的活性降低。同时纳米铝粉因较大的比表面积也极容易团聚成块导致在燃烧时出现燃烧不充分的现象,所以保持铝的活性和降低纳米铝粉的团聚现象成为现阶段一大研究热点。
通常解决此问题的主要方法是对铝粉表面进行包覆。鲁彦玲对铝粉包覆用的偶联剂进行了比较研究,但是未给出偶联剂与铝粉之间结合稳定性的定量数据。刘亚青等研究了HTPB在不同温度下与铝不同表面的结合能,得出在400 K温度下端羟基聚丁二烯(HTPB)与铝(011)面的结合能最大。Jiang等结合GCMC和MD模拟方法,研究了在不同pH值下在膜浇铸溶液中制备的聚多巴胺膜中水/丙烯的结构特性和吸附-扩散行为。王伯良等则通过建立不同乙酰柠檬酸三丁酯(ATBC)含量的PVDF/ATBC模型,模拟分析PVDF/ATBC体系的溶度参数、径向分布函数及玻璃化转变温度。朱宝忠等通过控制变量法,对硬酸酯包覆纳米铝粉的燃烧特性进行了研究,结果显示硬酸酯包覆的纳米铝粉比未包覆的纳米铝粉燃烧更充分,且有效的保持了纳米铝粉的活性。王军通过化学气相沉积法直接在n-Al表面聚合生成聚四氟乙烯(PTFE)层膜,放热量是物理混合的6倍,同时燃烧性能也得到显著改善。He等通过化学气相沉积法制作了n-Al@PDA/PTFE的亚稳态混合复合物材料(MIC),相比于与传统的n-Al/PTFE亚稳态复合材料,n-Al@PDA/PTFE表现出更高的能量释放和更低的灵敏度。最重要的是其可通过调节聚多巴胺的厚度来改变起始反应活化能,从而改变复合材料反应活性。陶俊利用MS软件模拟了聚四氟乙烯(PTFE)在不同温度下在氧化铝不同表面的结合能,得出在高温烧结状态下有利于PTFE在氧化铝中的均匀分布。
基于以上分析可知,以往研究绝大多数聚焦在使用单一材料对铝粉进行包覆或分子动力学研究,且主要关注的是铝粉包覆前后的活性变化,而忽视了包覆剂选择的理论依据和包覆剂吸附稳定性的评价,尤其是对不同材料吸附稳定性的对比分析。本研究拟通过分子动力学方法(MD),从微观角度出发,在相同条件下模拟研究不同包覆物在氧化铝表面的吸附特性,获得最佳的包覆物,以期为纳米铝粉包覆材料的选择提供理论依据。
2 MD模拟计算
2.1 氧化铝(0 0 1)面模型的构建
氧化铝超晶胞的构建采用Materials Studio 2020(MS)软件自带的晶体信息文件。其晶胞属于斜方六面体结构,晶胞参数为=0.476 nm,=4.579 nm,=12.591 nm。为了达到研究不同物质在氧化铝表面吸附的目的,在导入晶体信息文件之后,我们采用MS软件对其(0 0 1)面进行切割分面,采用Forcite模块在COMPASSⅢ立场下进行结构优化。值得注意的是氧化铝为离子型晶体,我们必须在分配力场之后删除其Al-O键,才能正确的进行后续的计算。为优化后的表面建立6×6超晶胞结构,添加0Å真空层将其转变为三维周期性结构。
2.2 力场的选择
COMPASS力场是第一个基于从头计算、能够准确预报孤立态和凝聚态分子的分子结构、构象、振动、热力学性质的分子力场。同时也是第一个把以往分别处理的有机分子体系的力场与无机分子体系的力场统一的分子力场。COMPASS力场的参数是根据实验值进行优化,并使用凝聚相分子的大量数据进行参数化。通过使用MD方法获得的液相和固相分子的热物理性质,进一步修正和验证了其非键合参数。采用COMPASS力场能够准确模拟有机物、无机物、聚合物,一些金属离子、金属氧化物与金属。尤其在处理有机与无机体系时,能够采用分类别处理的方式,不同的体系采用不同的模型,即使对于两类体系的混合,仍然能够采用合理的模型描述。为了更为准确的进行本模拟,MD模拟采用COMPASS力场的优化版本COMPASSⅢ力场。
2.3 MD参数设置
为模拟对照不同物质在氧化铝表面的吸附,将所有模型的动力学参数均设为相同。MD模拟采用COMPASSⅢ力场,温度设为298 K,初始速率选取Random,静电力(库仑力)和非键力中范德华力作用分别选用Ewald和Atom-based长程加和法,精度设置为Medium,程序会自动将球形截断半径设为12.5Å,电荷为Forcefield assigned,控温方法选择Andersen程序进行控温。模拟时间步长为1 fs,总模拟步数为20万步,总模拟时间为200 ps,前10万步用于体系平衡,后10万步用于统计分析。
2.4 吸附物/Al2O3(0 0 1)吸附模型的构建
目前聚多巴胺的聚合机理学术界还没有定论,当下通用的PDA聚合机理采用贺武研究的聚合理论。分子结构如图1所示,在苯环的3、6位置为PDA分子的聚合位置。据此,MD模型通过Amorphous模块构建了聚合度为10的聚多巴胺链,密度为1.3 g/cm。之后在Smart模式下对其进行了结构优化,用AC模块为其建立三维周期性结构。界面模型通过分层共聚的形式实现,采用MS软件的层工具建立聚多巴胺/AlO(0 0 1)层模型,为聚合物建立30Å的真空层结构,固定全部AlO分子。而后在正则系综(NVT)下进行20万步的动力学模拟,分析其结合能大小。聚多巴胺/AlO(0 0 1)吸附模型如图2(a)所示。

图1 聚多巴胺分子结构式
PVDF/AlO(0 0 1)吸附模型的构建方法和前面所述相同。PVDF密度设定为1.8 g/cm。PVDF/AlO(0 0 1)吸附模型如图2(b)所示。
HTPB/AlO(0 0 1)吸附模型的构建方法和前面所述相同。HTPB密度设定为0.908 g/cm。HTPB/AlO(0 0 1)吸附模型如图2(c)所示。
PTFE/AlO(0 0 1)吸附模型的构建方法和前面所述相同。PTFE密度设定为2.2 g/cm。PTFE/AlO(0 0 1)吸附模型如图2(d)所示。
硬酸酯/AlO(0 0 1)吸附模型的构建方法和前面所述相同。设定其密度为0.84 g/cm。唯一不同的是,由于硬酸酯没有可打开的双键供其进行聚合反应,所以在模拟体系中只放置了一个硬酸酯分子。PTFE/AlO(0 0 1)吸附模型如图2(e)所示。
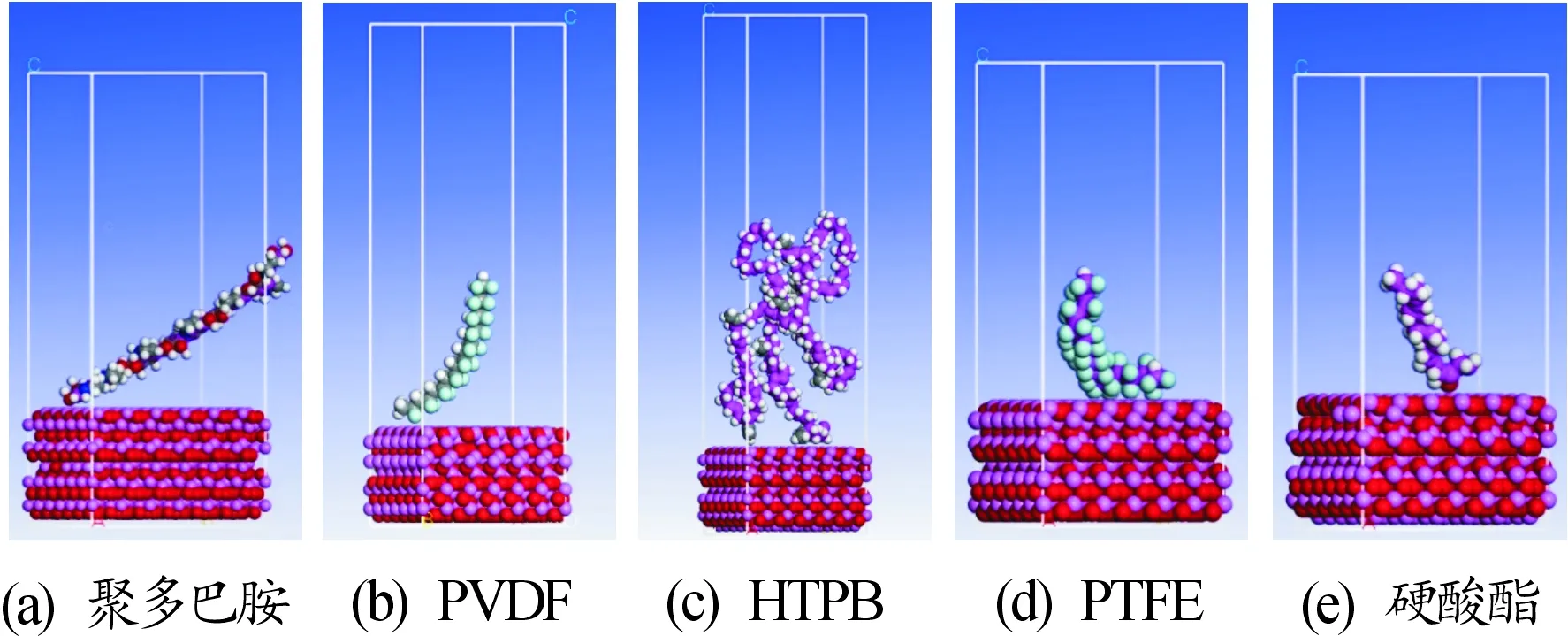
图2 经处理的包覆物吸附模型示意图
3 结果分析与讨论
3.1 模拟分析
从图2和图3的对比中可以直观的看到各种包覆物在进行MD模拟之后,均吸附在AlO表面。其中,聚多巴胺由直链状态变为弯曲状态,且聚合物链中的绝大部分都吸附在AlO表面。同时从图3中可以明显看到HTPB在整个模拟体系中所占的比例比聚多巴胺在模拟体系中所占的比例要高,而在模拟之后只有聚合物链下部的少量物质吸附在AlO表面,上部的聚合物链并没有看到有明显的团聚现象出现,这也说明HTPB物质自身没有特别强的自吸附能力。而PVDF和PTFE由于成链之后链节较短,硬酸酯没有发生聚合反应,所以构建共晶体系之后包覆物在体系中所占的含量相对较少。虽然由MD模拟完成之后,这3种物质均完全吸附在AlO表面,但无法从图中直观的判断哪种物质结合的稳定性更高。

图3 包覆物在Al2O3(0 0 1)面吸附后模型示意图
3.2 结合能分析
结合能是表征同一体系2种物质之间结合稳定性的重要参数,对混合体系的力学性能有重要影响。结合能的公式可表示为:
=-=-[-(+)]
式中:为反应体系的界面能,结合能为界面能的负值。当界面能为负值时说明体系吸附稳定。若为正值时说明包覆物存在吸附和解吸附的现象,吸附性能不稳定。通过计算,对应的5种包覆剂的结合能的大小如表1所示。
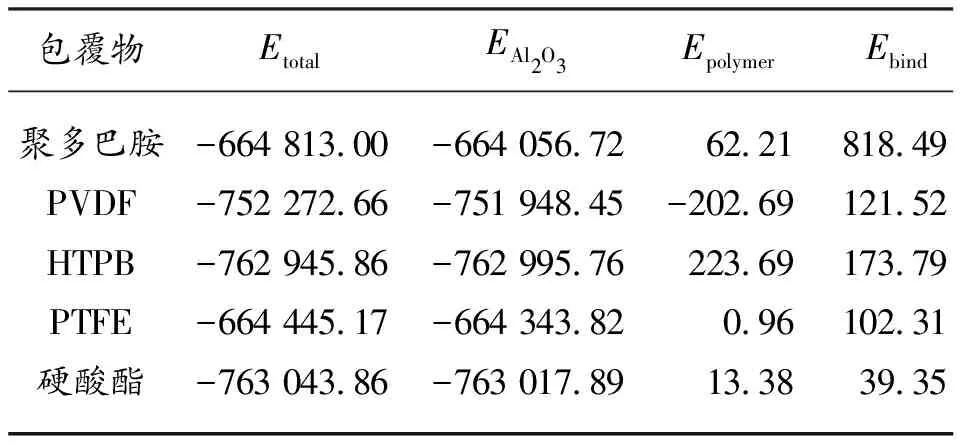
表1 不同包覆物与Al2O3(0 0 1)面的 结合能(kcal·mol-1)
由表1数据分析得到,所有包覆物的结合能均为正值,吸附稳定。其中聚多巴胺的结合能最高为818.49 kcal/mol。HTPB次之,结合能最低的是硬酸酯。造成硬酸酯结合能低的主要原因极有可能是其在模拟体系中所占含量较低导致的,加大浓度之后硬酸酯的结合能完全有可能增加。但是从聚多巴胺和HTPB的结合能分析得出,并不是包覆物的含量高其结合能就一定大,结合能的大小与包覆物本身的性质有极大的关系。由此可以确定在五种包覆物中聚多巴胺与AlO表面的结合能是最高的,其吸附稳定性也是最强的。将来可以通过聚多巴胺和结合能较低的包覆物进行联合使用,发挥联合优势制作核壳结构的包覆粒子,以达到改善n-Al颗粒的燃烧性能和贮存性能。
4 结论
1) 5种物质中聚多巴胺在氧化铝表面的结合能最大,可以作为交联剂使用。
2) 具有吸附性能的包覆物,并不是在体系中含量越高吸附性能越强,吸附性能和物质本身的性质有关。
3) MD模拟方法能够从结合能的角度对包覆剂的包覆效果进行评价,简单可行。
