铝掺杂四氧化三锰低温催化氧化气态甲苯
2022-05-19刘立忠姚慕舟汤艳峰丁建东
刘立忠,姚慕舟,汤艳峰,丁建东,张 彦
(南通大学 化学化工学院,江苏 南通 226019)
挥发性有机化合物(volatile organic compounds,VOCs)作为全球型大气污染物,是造成城市灰霾、光化学烟雾、大气毒性等复合型大气污染现象的主要推手之一,给人们的日常生活、健康带来严重影响[1-4]。当前,国内外许多学者致力于VOCs 污染治理技术的研究,传统的技术有吸附、冷凝、生物处理,新技术有低温等离子体、光催化氧化、膜分离等[5-7],但这些技术均未实现大规模工业化应用。与热解、吸附、吸收等处理手段相比,催化氧化方法因净化率高、耗能低等优点被认为是最有工业化应用前景的技术之一[8-9]。催化氧化技术的关键是催化剂,常用的催化氧化催化剂包括贵金属和过渡金属氧化物。贵金属催化剂具有活性高的优点,但其价格昂贵、高温易烧结、易积碳等缺点限制了其广泛应用[10-12]。相较于贵金属催化剂,过渡金属氧化物因来源广、成本低、多价态、结构可调性强等优势而成为催化领域研究的热点催化剂。而单金属氧化物的低温催化活性一般,且其形貌结构也会因长时间反应而发生改变,导致催化活性下降,其难以满足长期稳定使用的目的[13-14]。
针对此问题,学者们通常选择向纯相的MnOx中添加异质原子来提高和稳定其低温催化活性。Kim 等[15]合成了Mn3O4、Mn2O3和MnO2催化剂来催化氧化甲苯和苯,结果发现掺杂K、Ca 和Mg 的Mn3O4催化剂表现出了更高的催化活性。He 等[16]研究设计了KxMnO2的纳米球,并用来催化氧化HCHO,结果表明KxMnO2比MnO2、MnOx粉末具有更高的催化活性。本课题组前期将Sm、Fe、Cu 等异质原子掺杂到Mn3O4晶体结构中,发现异质原子掺杂可有效诱导催化剂产生更多的表面缺陷和氧空位,提高了催化剂的低温催化反应活性[17-18]。可见,不同异质原子掺杂催化剂会导致催化剂结构及性能的差异。因此,继续研究开发不同异质原子掺杂催化剂依然是进一步推进VOCs 低温催化反应活性的有效途径。
本文以Mn3O4作为锰基催化剂的代表并以Al作为异质原子,通过一步熔融聚合法制备了掺杂不同Al 含量的Al:Mn3O4复合催化剂,探究了Al 掺杂对催化剂催化氧化甲苯性能的影响,考察了反应气氛及反应时间对掺杂催化剂稳定性的影响,探讨了催化剂催化氧化甲苯的反应机理。
1 实验部分
1.1 实验药品
Mn(CH3COO)2·4H2O,分析纯,南通启翔生物科技有限公司;Al(NO3)3·9H2O,分析纯,南通启翔生物科技有限公司;柠檬酸(C6H8O7·H2O),分析纯,南通启翔生物科技有限公司;甲苯(C7H8),分析纯,国药集团化学试剂有限公司。
1.2 实验仪器及测试条件
X 射线衍射(岛津,XRD-7000)用于分析催化剂的晶体结构,测试条件为:Cu 靶Kα 辐射源(λ=0.154 06 nm),扫描速度2 (°)/min,扫描范围10°~70°,电压40 kV。双通道气相色谱(福立,GC-9790),配有甲烷转化炉和双FID 检测器,用于测定甲苯浓度和二氧化碳浓度。
1.3 Al:Mn3O4 催化剂的制备
本实验采用熔融法[19]制备Al:Mn3O4复合催化剂,根据Mn 与Al 的摩尔比标记为Al:Mn3O4-x(x=5,4,3,2),分别对应于Al:Mn3O4-5、Al:Mn3O4-4、Al:Mn3O4-3、Al:Mn3O4-2 4 种复合氧化物。以Al:Mn3O4-4 的制备过程为例,具体操作步骤如下:称取1.961 g Mn(CH3COO)2·4H2O 固 体、0.750 g Al(NO3)3·9H2O固体和2.882 g 柠檬酸,混合研磨至黏糊状。将黏糊置于马弗炉中焙烧(5 ℃/min),先在200 ℃空气氛围中停留2 h,再在450 ℃空气氛围中停留3 h,得到Al:Mn3O4-4 复合氧化物,最后将其研磨成粉末状,装样备用。以实验设定比例的Mn(CH3COO)2·4H2O、Al(NO3)3·9H2O 和柠檬酸为原料,用同样的方法制备复合氧化物。为了进行对比实验,Al2O3和Mn3O4也均采取上述类似的方法制备。
1.4 催化剂的催化性能测试
本实验采用甲苯作为模拟VOCs 污染物评估催化剂的催化活性。将需要评估的催化剂装入固定床催化反应管中部并固定于管式炉加热槽中,设定管式炉升温至预定值。通过质量流量计控制系统总气体流量为100 mL·min-1,设置空速为vWHS=30 000 mL·g-1·h-1,并调节进气流量使进口处的甲苯质量浓度约为1 000 mg·m-3,以及使系统湿度达到预定值。在对所有催化剂进行评价前,我们已进行无催化剂的空白实验并确认350 ℃下无VOC 转化发生,同时也排除了催化体系的内外扩散因素的影响。VOC 的转化率(αVOC)、CO2收率(YCO2),以及VOC总氧化和CO2产生的表观活化能Ea(VOC)和Ea(CO2)的计算公式为
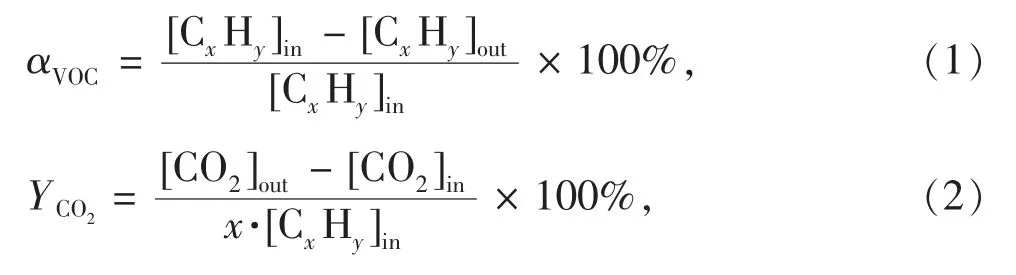
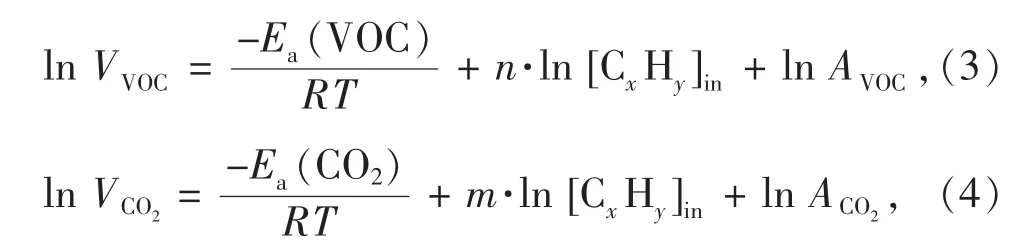
式中:[CxHy]in和[CxHy]out分别为进、出口的VOC 浓度,单位为mol·m-3;[CO2]in和[CO2]out分别为进、出口的CO2浓度,单位为mol·m-3;VVOC为VOC 的总氧化速率,VCO2为转化成CO2部分的VOC 氧化速率,单位为mol·m-3·s-1;n 和m 分别为VOC 氧化和CO2收率(矿化部分的VOC)的表观反应常数;R 为气体常数;T 为反应温度;A 为指前因子。
2 结果与讨论
2.1 催化剂结构分析
低离子半径的异质离子易进入催化剂体相中破坏催化剂的晶体结构和促进催化剂使用时表面产生更多的氧空位[18,20]。为了证实Al 掺杂能促进Mn3O4使用时产生氧缺陷,通过XRD 对Mn3O4和Al:Mn3O4-4 氧化物催化剂的晶体结构进行表征,结果如图1 所示。从图中可以看出,单纯醋酸锰和柠檬酸的煅烧所形成的锰氧化物在2θ=18.02°、28.97°、31.03°、32.41°、36.04°、44.37°、53.89°、60.02°、64.68°处的衍射峰均能够与Mn3O4氧化物的(101)、(112)、(200)、(103)、(211)、(220)、(312)、(224)和(314)晶面(JCPDS No.18-0803)相对应,且形成的Mn3O4峰形尖锐,表明结晶度较好[18]。此外,将Al 盐、Mn 盐和柠檬酸混合煅烧后获得样品的XRD 衍射峰也均与Mn3O4衍射峰一致,且没有出现其他明显的杂质衍射峰,说明样品Al:Mn3O4-4 与Mn3O4有相似的晶体结构。同时也发现Al:Mn3O4-4 的XRD衍射峰出现了明显的宽化现象,说明适量小离子半径Al3+(rAl3+=0.053 5 nm)掺杂占据了离子半径相对较大的Mn3+(rMn3+=0.058 0 nm)的位置,并导致了单元晶胞的异向伸缩,破坏了Mn3O4晶体结构,有利于活性氧从氧空位中溢出,促进甲苯氧化[21-24]。
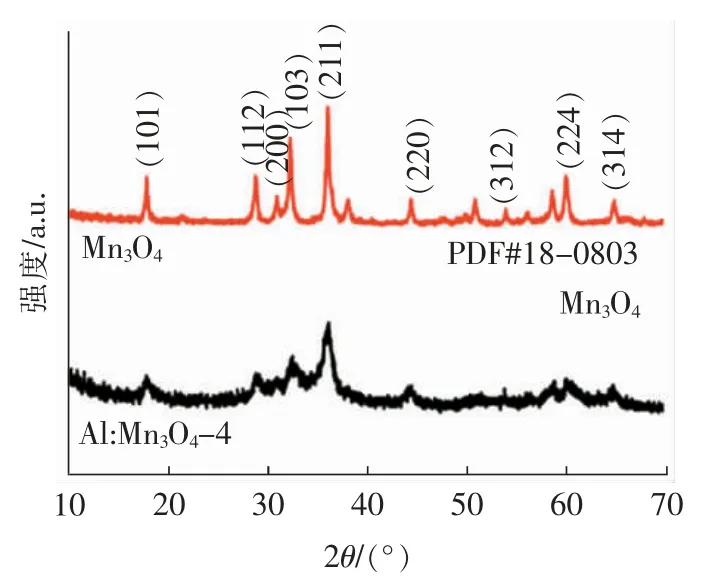
图1 Al:Mn3O4-4 和Mn3O4 催化剂的XRD 图Fig.1 XRD patterns of Al:Mn3O4-4 and Mn3O4
2.2 不同Mn/Al 比对催化活性的影响
图2 展示了不同催化剂催化氧化甲苯的转化率与反应温度间的变化趋势曲线图。从图中可以看出,各种催化剂的总氧化活性均随着反应温度的升高而提升,说明较高的反应温度有助于激活催化剂产生更多的表面活性氧。另外也可以看出,6 种样品催化氧化甲苯的曲线在230 ℃前后存在不一样的变化趋势。小于230 ℃时,样品总氧化活性的递降顺序如下:Al:Mn3O4-4 >Al:Mn3O4-3 >Al:Mn3O4-5 >Mn3O4>Al:Mn3O4-2 >Al2O3,即Al:Mn3O4-4 的低温催化氧化活性最佳。而当反应温度大于230 ℃时,Mn3O4的总氧化甲苯的活性迅速提升,且240 ℃后活性已明显高于其他样品。该现象表明:反应温度小于230 ℃时Mn3O4的表面活性氧与锰离子之间的化学键很难断裂,适量铝掺杂导致催化剂结构松散,促进了表面活性氧与锰离子之间化学键的断裂。为了更好地比较不同催化剂的催化活性,采用转化率为50%和90%时的反应温度T50%和T90%进一步评估催化剂的催化能。从表1 中可以看到,Al:Mn3O4-5 催化氧化甲苯转化的T50%和T90%分别为245 和273 ℃,Al:Mn3O4-4 的T50%和T90%分别为230和278 ℃,Al:Mn3O4-3 的T50%和T90%分别为240 和257 ℃,Al:Mn3O4-2 的T50%和T90%分 别 为278 和297 ℃,Mn3O4的T50%和T90%分别为235 和249 ℃。然而,氧空位的循环再生需要Mn3+↔Mn2+和Mn4+↔Mn3+之间的良好转化,由于Al 的价态通常为+3 价,且单独氧化铝的催化氧化活性较差(见图2),所以当掺入微量的铝时(n(Mn)/n(Al)=5),Al 取代造成的催化剂结构变化对活性的影响低于失去活性锰的作用影响;同时,过多的铝掺入也会导致活性锰的大量消失,也不利Mn3O4催化活性的提升。
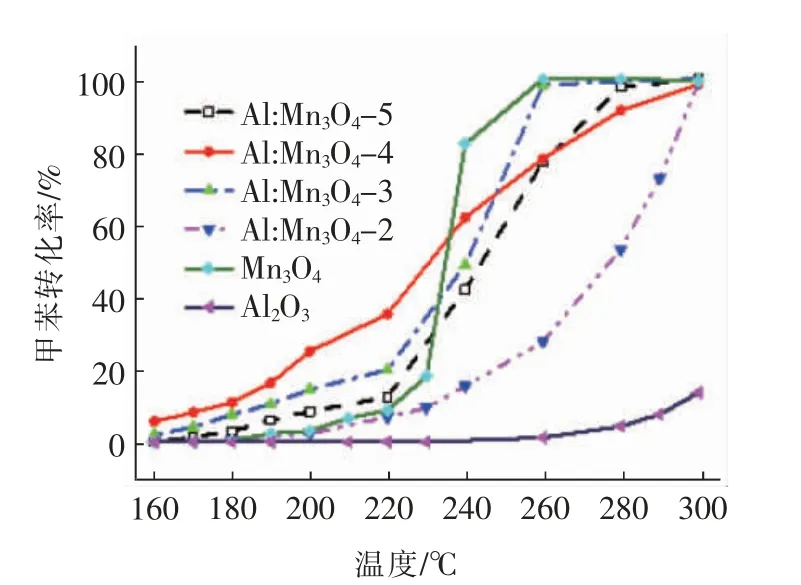
图2 不同催化剂总催化氧化甲苯的性能Fig.2 Total catalytic oxidation of toluene over different catalysts
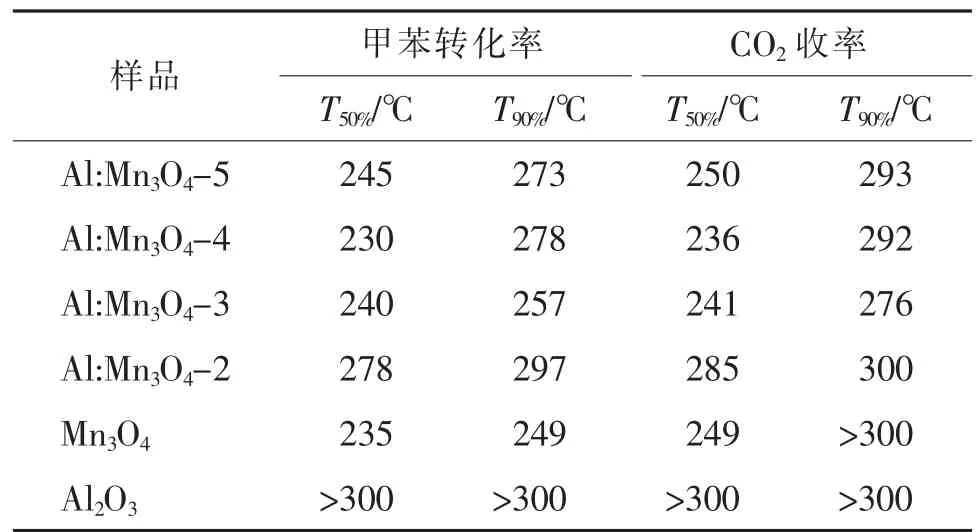
表1 不同催化剂的催化活性比较Tab.1 Comparison of catalytic activities over different catalysts
图3 为上述6 种催化剂催化氧化甲苯时的CO2收率变化曲线。由图可知,低温反应的曲线分布趋势与图2 中的甲苯转化率的类似,也是Al:Mn3O4-4催化剂的CO2收率最高,但较高温度区间的走势则略有不同。很明显,对于纯Mn3O4催化剂,其在280℃后的CO2收率基本稳定在81%,造成这一现象的可能原因是较高的反应温度导致Mn3O4催化剂表面结构发生了变化,其表面的活性氧物种浓度不足以对如此浓度下的甲苯进行彻底的矿化。另外,将表1中各Al:Mn3O4催化剂获得CO2收率T50%和T90%分别与相应催化剂催化氧化甲苯的转化率数据对比,可发现所有催化剂的CO2收率T50%和T90%均要高于甲苯转化率的。该现象表明,催化剂在发生催化氧化反应过程中存在副产物。事实上,大多数锰基氧化物在低温催化氧化过程中,由于较低反应温度不足以驱动催化剂表面产生足够多的活性氧去矿化VOCs 分解的中间产物,所以催化反应后的尾气中会残留一定量的有机副产物,如苯甲醛、苯甲酸、2(5H)-呋喃酮、柠檬酸、乙酸和丙酮等[25-26]。
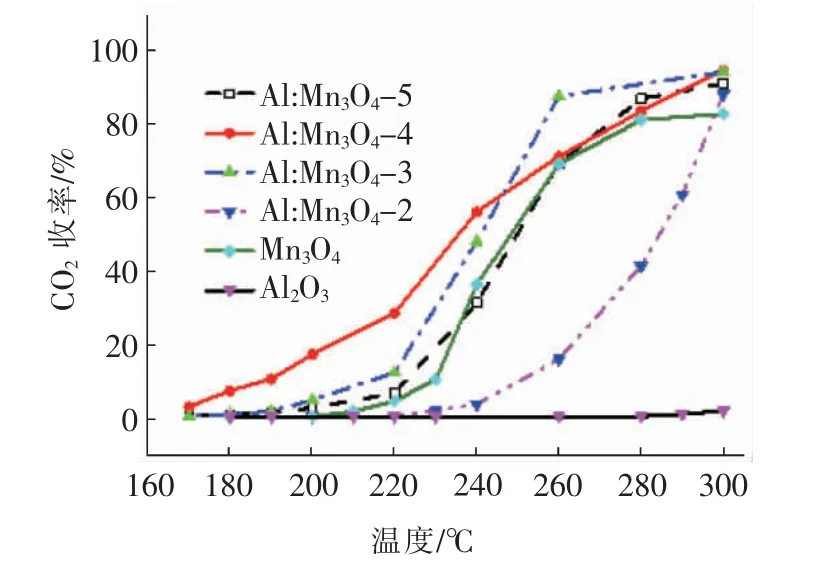
图3 不同催化剂催化矿化甲苯的性能Fig.3 Catalytic mineralization of toluene over different catalysts
2.3 催化反应动力学研究
根据式(3)和式(4)及图2 和图3 中Mn3O4和Al:Mn3O4-4 催化剂在转化率低于20%情形下的甲苯转化效率和CO2收率的数据,可获得Mn3O4和Al:Mn3O4-4 催化剂的催化反应表观活化能。从图4中可以看到,两种催化剂催化氧化甲苯反应的ln V相对1 000/T 均呈现良好的线性关系,通过推导计算可得两种催化剂的表观活化能值。Mn3O4催化氧化甲苯的总氧化表观活化能Ea(VOC)值为97.94 kJ/mol,对应于CO2收率部分的表观活化能Ea(CO2)值为144.23 kJ/mol。很明显,Ea(CO2)值要远大于Ea(VOC)值,该结果也说明了催化剂催化氧化甲苯的反应存在副反应,的确有部分甲苯没有被完全矿化为CO2,这与图2 的甲苯总氧化和图3 的CO2收率之间的差异结果分析相一致。此外,从图4 中也可以看到,当向Mn3O4中掺入摩尔分数为20%的Al 后,催化剂(Al:Mn3O4-4)的两种表观活化能值均出现了降低:Ea(VOC)值为58.06 kJ/mol,Ea(CO2)值为106.35 kJ/mol。这一结果可归因于适量Al 掺杂造成了Mn3O4催化剂结构的适度松散,使得催化剂表面在热作用下更容易形成氧缺陷,从而有利于更多表面吸附氧的不断循环再生,推进甲苯的低温催化氧化反应。此外,Al:Mn3O4-4 催化氧化甲苯的表观反应活化能值(58.06 kJ/mol)要好于文献报道的Ni0.5Zn0.5Fe2O4(94.00 kJ/mol)[27]、3DOM LaMnO3(66.00 kJ/mol)[28]、30%MnOx/Al2O3(124.00 kJ/mol)[29]、GC-LaMnO3(84.00 kJ/mol)[30]、3D-Eu0.6Sr0.4FeO3(78.80 kJ/mol)[31]、La0.6Sr0.4Fe0.8Bi0.2O3-δ(127.70 kJ/mol)[32]等催化剂的。
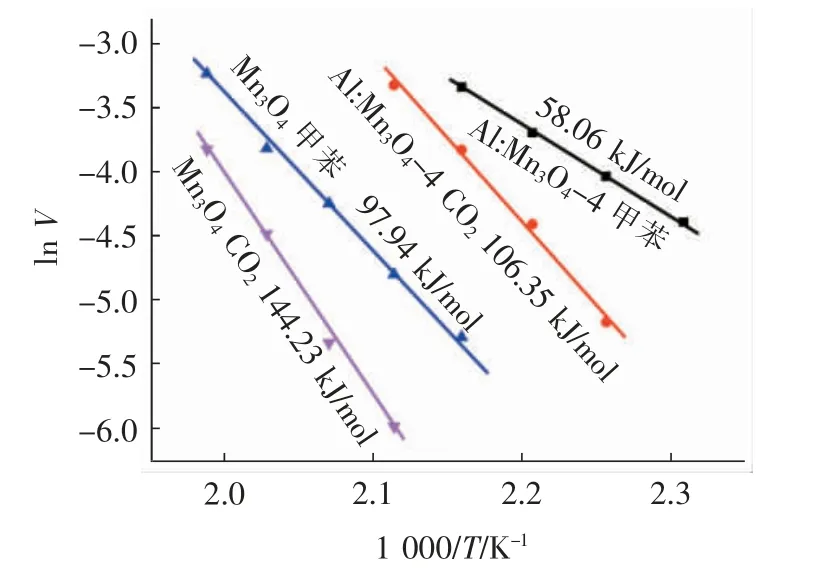
图4 Mn3O4 和Al:Mn3O4-4 催化剂催化氧化甲苯的总氧化和氧化为CO2 部分的阿伦尼布斯曲线Fig.4 Arrhenius curve for toluene conversion and CO2 yield over Mn3O4 and Al:Mn3O4-4
2.4 水汽含量对催化反应活性的影响
图5 为不同水蒸气通入量对Al:Mn3O4-4 催化剂催化氧化甲苯的活性影响曲线。水蒸气通入量按照水路流量(Qwater)与总流量(Qtatol)的比值进行调节,比值分别为0%、5%、10%、25%和45%。从图5曲线中可以看出,通入少量的水蒸气(5%)对催化剂总催化氧化甲苯活性影响较小。众所周知,甲苯的总氧化转化反应只需发生第1 步甲基氧化反应即可,所以少量水蒸气的引入对气氛中的氧气含量影响较小,不足以引起较大的氧化转化率的变化[33]。而当水蒸气流量占比位于5%~45%区间时,甲苯的催化氧化活性明显被削弱。一方面,大量的水分子会降低反应气氛的氧含量;另一方面,水分子会与氧气分子竞争催化剂表面的氧空位,过多水分子侵占氧空位,显然会抑制氧气在氧空位处分解,进而阻碍了活性氧的循环再生过程,极大降低了甲苯的氧化效率。
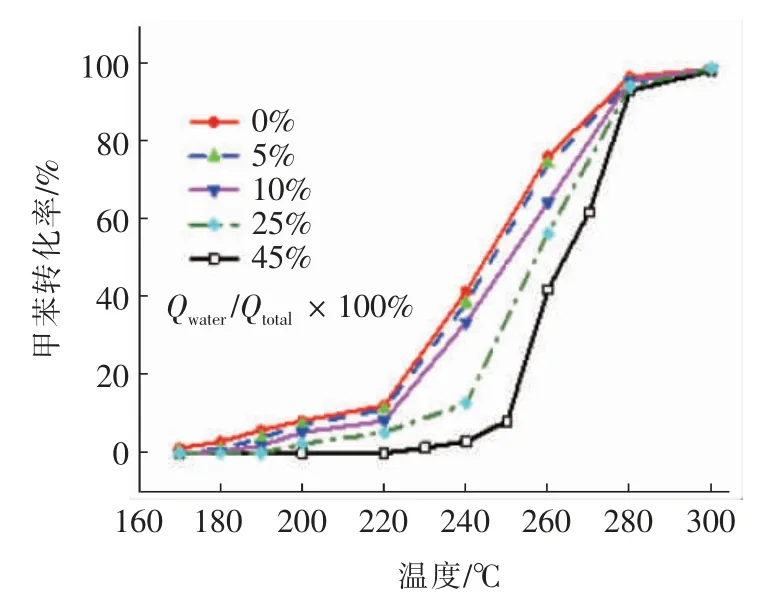
图5 水蒸气通入量对Al:Mn3O4-4 催化剂催化氧化甲苯活性的影响Fig.5 Effect of water vapor on toluene oxidation over Al:Mn3O4-4
2.5 催化稳定性测试
根据上述实验分析结果可知,Al:Mn3O4-4 具有更好的低温催化氧化甲苯活性。为了进一步考察催化剂的低温催化活性的稳定性,分别将Al:Mn3O4-4 和Mn3O4催化剂的用量调至原来的5 倍,在200 ℃下对其进行约6 h 的稳定性测试,结果如图6 所示。从图中可以看到,增加用量的催化剂在200 ℃下的反应活性明显高于原催化剂的催化氧化活性,说明增加催化剂用量显然可以提高催化反应过程中的总表面活性氧浓度。同时也可发现,Al:Mn3O4-4 的低温催化氧化活性的稳定程度要明显高于Mn3O4催化剂,在6 h 的恒温测试过程中,Al:Mn3O4-4 催化剂保持了较稳定的催化反应活性,而Mn3O4催化剂在低温反应2 h 后活性出现明显衰减。这一现象表明,低温反应可能会造成Mn3O4表面活性氧数量的不足,导致其表面逐渐积碳结焦,进而阻碍活性氧对甲苯的氧化进攻,要维持其稳定的催化氧化反应活性需要升高反应温度或继续加大催化剂的用量。对于Al:Mn3O4-4 催化剂,由于适量Al 在Mn3O4晶体结构中的掺杂诱导,促进了复合催化剂表面更容易产生活性氧,有利于增加其同等低温条件下的活性氧数量和抑制反应中间产物在催化剂表面的积累,从而使得其催化活性得以保持。
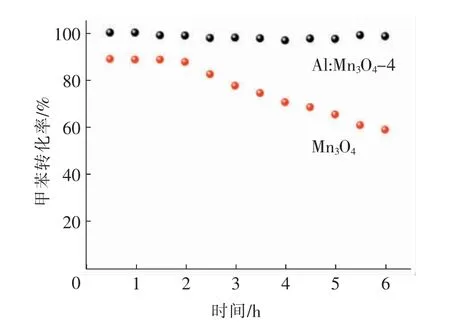
图6 低温下Al:Mn3O4-4 和Mn3O4 催化剂长时间催化氧化甲苯的活性曲线(200 ℃)Fig.6 Low-temperature catalytic oxidation of toluene over Al:Mn3O4-4 and Mn3O4 for a long time(200 ℃)
2.6 反应机理探讨
依据上述表征结果及性能测试,推测Al:Mn3O4-4 催化氧化甲苯的反应机理,如图7 所示。催化氧化反应的本质是活性氧的进攻消耗与再生循环过程,因此,催化剂表面活性氧数量决定了催化反应的程度[19]。由在常态下催化剂表面的吸附氧物种(Oads:化学吸附在催化剂表面氧空位中,并提供给Mn4+或Mn3+1 个电子形成Mn3+或Mn2+。当催化剂被逐渐升温到一定温度后,吸附氧被激发并带走电子形成活泼的氧负离子同时留下氧空位,这将导致对应的锰价态从Mn3+转变成Mn4+和Mn2+转变成Mn3+,溢出的吸附氧Oads迅速去进攻甲苯中甲基碳原子,将甲苯连续氧化成苯甲醇→苯甲醛→苯甲酸→…→直链有机物→二氧化碳[18,25]。消耗掉的吸附氧则由氧空位从气相中捕捉氧气分子并将其分解转化为吸附氧来补充,外部氧气的不断补给促成了整个催化氧化反应的持续进行。
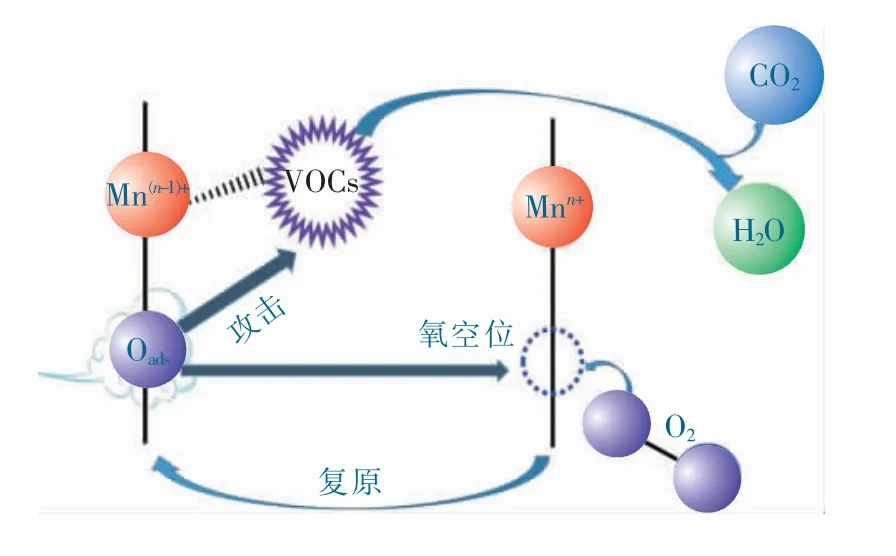
图7 催化剂Al:Mn3O4-4 催化氧化VOCs 的过程示意图Fig.7 Schematic of VOCs oxidation over Al:Mn3O4-4
3 结论
本文通过熔融聚合法制备了一系列铝掺杂的Mn3O4催化剂,并以甲苯作为模拟VOCs 污染物,评价了各种催化剂的反应活性。研究结果表明,适量Al 在Mn3O4中掺杂破坏了催化剂晶体结构,有利于表面活性氧从氧空位中溢出,促进催化剂的低温催化氧化活性。在所有催化剂中,Al:Mn3O4-4 催化剂取得了最高的低温催化氧化甲苯活性,其转化率在50%的反应温度(230 ℃)比Mn3O4的(235 ℃)向低温推进了5 ℃。同时,Al:Mn3O4-4 的总氧化和氧化为CO2部分的反应表观活化能(Ea(VOC)=58.06 kJ/mol,Ea(CO2)=106.35 kJ/mol)均低于Mn3O4的数值。低含量(小于5%)的水蒸气对Al:Mn3O4-4催化氧化甲苯转化反应影响较低,高水蒸气量由于会过度压缩反应气氛中氧气的占比和抢占催化剂表面的氧空位而导致催化剂催化转化甲苯的能力出现明显下降。此外,Al:Mn3O4-4 催化剂在200 ℃下经过6 h 的催化氧化反应活性依然稳定。
