肺动脉高压孤儿药Selexipag合成工艺的优化
2022-05-05周鹏飞尹传奇
周鹏飞,鄢 龙,王 刚,尹传奇
武汉工程大学化学与环境工程学院,湖北武汉 430205
Selexipag,化学名2-{4-[N-(5,6-二苯基吡嗪-2-基)-N-异丙基氨基]丁氧基}-N-(甲基磺酰基)乙酰胺,是一种高度选择性的长效非前列腺素类前列 环 素 受 体 激 动 剂[1-4],由Actelion Pharmaceuticals Ltd和Nippon Shinyaku公司合作开发,并于2014年在日本获得了用于治疗肺动脉高压(plumonary arterial hypertension,PAH)的孤儿药称号,2015年12月21日口服selexipag片剂在美国获准用于治疗PAH,以延迟疾病进展并降低PAH住院治疗的风险[5]。2016年,肺动脉高压新药Uptravi(Selexipag商品名)获得欧盟委员会批准,用于世界卫生组织(world health organization,WHO)分级为Ⅱ-Ⅲ级的肺动脉高压患者的长期治疗。2020年10月,杨森制药向美国FDA提交了Uptravi静脉制剂的新药上市申请,用于治疗目前已处方口服疗法但暂时无法接受口服疗法、WHO功能分级Ⅱ-Ⅲ的肺动脉高压(PHA,WHO组I)成人患者。近期研究发现,selexipag与内皮素受体拮抗剂和/或5型磷酸二酯酶抑制剂联用,在治疗艾森曼格综合症(成人先天性心脏病中最严重的肺动脉高压形式)中有很好的效果[6]。据报道,在肺动脉高压前列环素受体激动剂肺动脉高压研究中,selexipag与其他以前列环素为途径的靶向药物相比较,具有更强的舒张血管效应[7]。
尽管Selexipag已成为肺动脉高压的治疗药物,但其合成方法仍需进一步优化,以降低成本和符合环保要求。樊等[8]以5-氯-2,3-二苯基吡嗪为原料,与中间体2-(4-氯丁氧基)四氢-2H-吡喃和2-氯-N-(甲基磺酰基)乙酰胺合成目标产物,该路线中有两步反应使用NaH,不适于放大生产。尚等[9]报道了4-((5,6-二苯基-2-基)(异丙基)氨基)-1-丁醇与2-氯-N-(甲基磺酰基)乙酰胺反应合成Selexipag的工艺,收率为80%。李兴民[10]以4-[(叔丁氧基羰基)(异丙基)氨基]-1-丁醇为原料合成2-[4-(异丙基氨基)丁氧基]-N-甲磺酰基乙酰胺,再与5-氯-2,3-二苯基吡嗪反应合成目标产物,总收率约为65%。浅木哲夫等[11]以5-氯-2,3-二苯基吡嗪为原料,经氯原子的氨基取代、醚键的生成、酯基水解及酰胺化得到目标产物,总收率为27%,相较于前者的合成方法,该路线虽然收率低,但工艺简单,易于工业化生产。本研究拟在浅木哲夫等合成方法基础上,以二苯基乙二酮为起始原料合成Selexipag,旨在优化反应步骤和探讨反应的最佳工艺条件,提高收率,为工业化生产奠定基础。合成路线如图1。
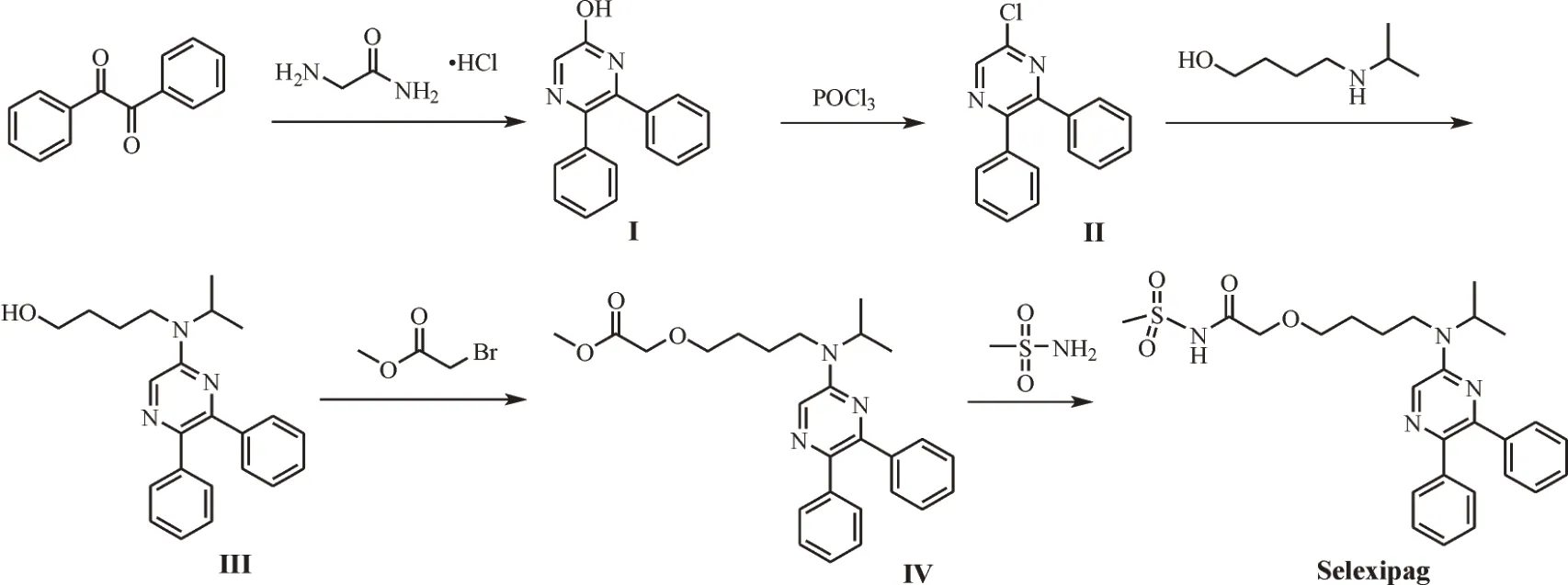
图1 Selexipage合成路线Fig.1 Synthetic process of Selexipage
1 实验部分
1.1 试剂与仪器
三氯氧磷(化学纯,国药集团化学试剂有限公司);二苯基乙二酮,氨基乙酰胺盐酸盐,4-异丙氨基丁醇,溴乙酸甲酯,甲基磺酰胺,氢氧化钠(分析纯,国药集团化学试剂有限公司);N,N-二甲基甲酰胺(N,N-Dimethylformamide,DMF),二氯甲烷,甲苯(分析纯,武汉格奥化学技术有限公司);叔丁醇钠,甲醇(分析纯,天津福晨化学试剂有限公司)。DPX 300核磁共振仪(德国Bruker公司),工作频率为400 MHZ;5975C型GC-MS仪(美国安捷伦科技有限公司);岛津LC-20AT型高效液相色 谱(high performance liquid chromatography,HPLC)(日本岛津公司)。
1.2 实验过程
1.2.1 5,6-二苯基吡嗪-2-醇(Ⅰ)和5-氯-2,3-二苯基吡嗪(Ⅱ)的合成化合物Ⅰ按文献[12]方法合成:将210.0 g二苯基乙二酮(1.0 mol)和110.0 g氨基乙酰胺盐酸盐(1.0 mol)加入到0.167 L 12 mol/L NaOH(2.0 mol)和1.5 L CH3OH中反应。处理后得235.0 g白色固体(HPLC测定质量分数96.99%)。ESI-MSm/z:248.32。
将上述白色固体加入到1 L DMF中,搅拌,缓慢加入0.18 L POCl3(1.7 mol),反应液由黄色逐渐变为棕褐色。滴加完毕后,加热到120℃反应4 h。冷却,将反应混合物倒入1.5 L水中,过滤,滤饼用水淋洗。将滤饼和0.5 L水混合,滴加饱和NaHCO3溶液至pH=7~8,过滤得到248.6.0 g淡黄色粉末(HPLC测定质量分数:97.15%)。由原料到化合物Ⅱ的产率为93.2%。1H NMR(400 MHz,CDCl3)δ8.60(s,1H),8.03(d,J=7.5 Hz,2H),7.59(m,2H),7.32(m,5H)。
1.2.2 4-((5,6-二苯基-2-基)(异丙基)氨基)-1-丁醇(Ⅲ)的合成将212.0 g化合物Ⅱ(0.82 mol)和0.50 L 4-异丙氨基丁醇(3.28 mol)加入到1 L三口反应瓶中,搅拌,升温至170℃反应68 h。反应完毕,浓缩,将残余的褐色油状粘稠物投入到1 L的H2O/DCM(体积比1/1)混合液中,搅拌30 min,分液,水相用DCM萃取1次。合并有机相,无水硫酸镁干燥,浓缩。将粗品用CH3OH/H2O(体积比3/1)溶液重结晶,水洗,干燥得黄褐色粉末260.0 g,收率89.4%(HPLC测定质量分数97.79%)。ESI-MSm/z:362.25。
1.2.3 2-{4-[N-(5,6-二苯基吡嗪-2-基)-N-异丙基氨基]丁氧基}乙酸甲酯(Ⅳ)的合成将243.0 g化合物Ⅲ(0.67 mol)溶于2 L甲苯中,加入0.6 L质量分数45%NaOH溶液和23.0 g四丁基溴化铵(0.067 mol),搅拌。将反应温度维持在0~5℃,缓慢滴加0.063 L溴乙酸甲酯(0.67 mol)。滴加完毕,80℃下反应5 h。分出水相,有机相用0.5 L水洗涤、无水MgSO4干燥,减压浓缩得黄色液体289.0 g(HPLC测定质量分数98.21%),可直接用于下一步反应。1H NMR(400 MHz,CDCl3)δ8.40(s,1H),7.94(d,J=7.5 Hz,2H),7.54(dd,J=7.5 Hz,2H),7.21(dd,J=7.1 Hz,6H),4.32(s,2H),3.72(s,3H),3.54(t,2H),3.40(t,2H),2.97(m,1H),1.49-1.50(m,4H),1.24(d,J=6.5 Hz,6H)。ESI-MSm/z:433.61。
1.2.4 Selexipag的合成将289.0 g化合物Ⅳ(0.68 mol)、92.7 g叔丁醇钠(0.85 mol,0.96 mol)和278.9 g甲基磺酰胺(0.65 mol)加入到反应瓶中,搅拌,室温反应2 h。分批加入2 L水,继续搅拌1 h。抽滤,水洗(300 mL×3),得黄褐色稠状固体,用异丙醇重结晶得黄色粉末297.9 g,收率92.3%(HPLC测定质量分数98.94%)。由化合物Ⅲ到Selexipag的收率为89.5%。1HNMR(400 MHz,CDCl3)δ7.94(s,1H),7.38(d,J=7.2 Hz,2H),7.29(d,J=6.3 Hz,2H),7.21(dd,J=7.1 Hz,6H),4.72(s,1H),3.93(s,2H),3.54(s,2H),3.40(s,2H),3.23(s,3H),1.68(s,4H),1.24(d,J=6.5 Hz,6H)。ESI-MSm/z:496.63。
2 结果与讨论
2.1 2-羟基-5,6-二苯基吡嗪(Ⅰ)的合成
Kaftory[13]将氢氧化钠固体加入到回流的含有1,2-二苯基乙二酮与甘氨酰胺盐酸盐的甲醇中,反应1 h后中和得到二者的缩合产物Ⅰ,收率为60%。本研究将氢氧化钠水溶液和1,2-二苯基乙二酮与甘氨酰胺盐酸盐的甲醇溶液混合,回流4 h后中和,降温至10℃左右,析出化合物Ⅰ,收率达到95%。虽然后者反应时间延长,但操作简单安全,收率大大提高。
2.2 5-氯-2,3-二苯基吡嗪(Ⅱ)的合成
以吡啶、DIPEA或N,N-二甲氨基吡啶为缚酸剂,将POCl3作为溶剂回流可将吡嗪环上的羟基转化为Cl原子[14-16]。这种方法不仅POCl3用量大,且易腐蚀设备。何等[17]在有机溶剂环境中加入缚酸剂,将化合物Ⅰ与POCl3反应,得到化合物Ⅱ,减少了POCl3的使用量。本课题组前期工作发现[18],由于化合物Ⅰ的吡嗪环上有2个呈碱性的氮原子,不需额外加入缚酸剂,以DMF为溶剂即可与POCl3反应得到化合物Ⅱ。该方法大大减少了POCl3用量,省去了后处理时萃取和浓缩过程,使生产操作更简单。本研究通过三因子四水平(表1)正交实验进一步考察了化合物Ⅰ与POCl3的摩尔比、反应温度和时间对羟基氯代反应的影响,优化了反应条件,结果见表2。
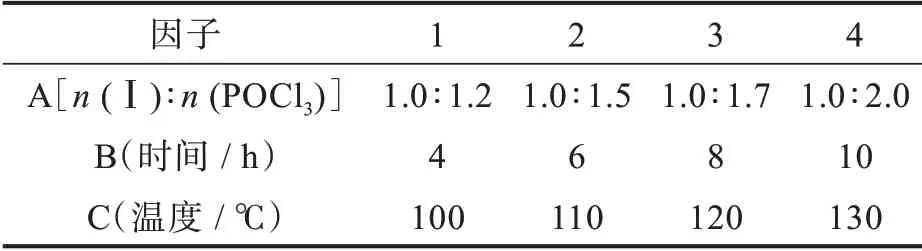
表1 羟基氯代反应的正交实验方案Tab.1 Orthogonal experimental scheme of hydroxyl chlorination reaction
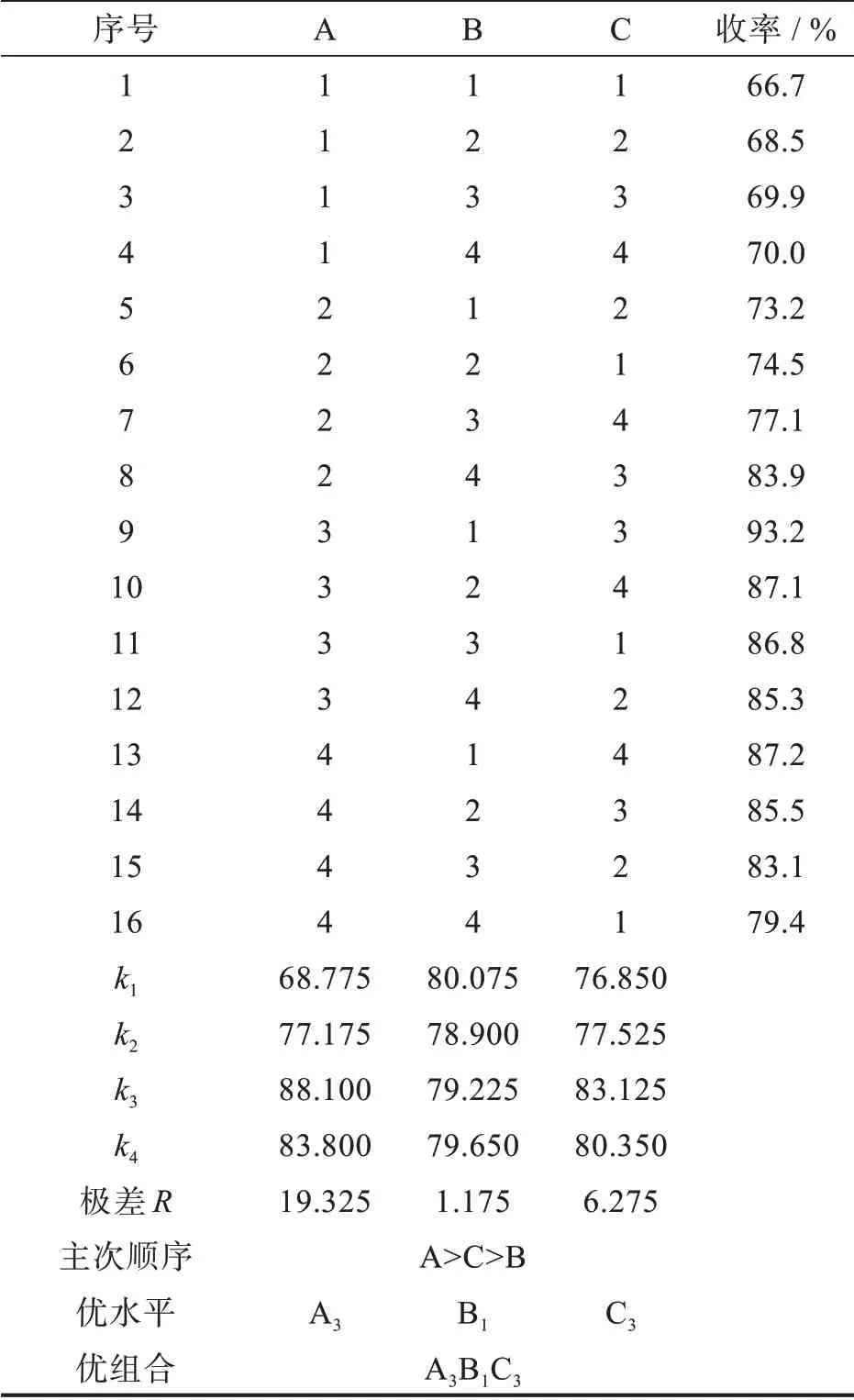
表2 氯代反应的正交实验结果Tab.2 Orthogonal experimental results of chlorination reaction
从计算出的K值得出A3、B1、C3最佳。通过公式计算出本实验中各Rj的值得到:R1>R3>R2,表明各因子A、C、B对氯代反应的影响逐渐减小。因此由化合物I与POCl3反应合成化合物Ⅱ的最优条件为:A3B1C3,即n(Ⅰ)∶n(POCl3)=1.0∶1.7,DMF溶剂中120℃反应4 h。反应完成后,直接加水析出固体,固体中和后即得化合物Ⅱ。
2.3 4-((5,6-二苯基-2-基)(异丙基)氨基)-1-丁醇(Ⅲ)的合成
4-异丙氨基丁醇同时具有-NH2和-OH基团,它与化合物Ⅱ发生氨基化反应时,应避免-OH的竞争性反应。前期研究发现,在4-异丙氨基丁醇作为氨基化试剂与化合物Ⅱ发生反应时,还可作为溶剂和缚酸剂,过量的4-异丙氨基丁醇减压浓缩回收,避免了其它合成方法后处理时大量废水的产生[18]。本研究进一步考察了化合物Ⅱ与4-异丙氨基丁醇的摩尔比、反应温度和时间对氨基化反应的影响,通过三因子四水平(表3)正交试验优化了反应条件,结果见表4。

表3 氨基化反应正交实验方案Tab.3 Orthogonal experimental scheme for amination reaction
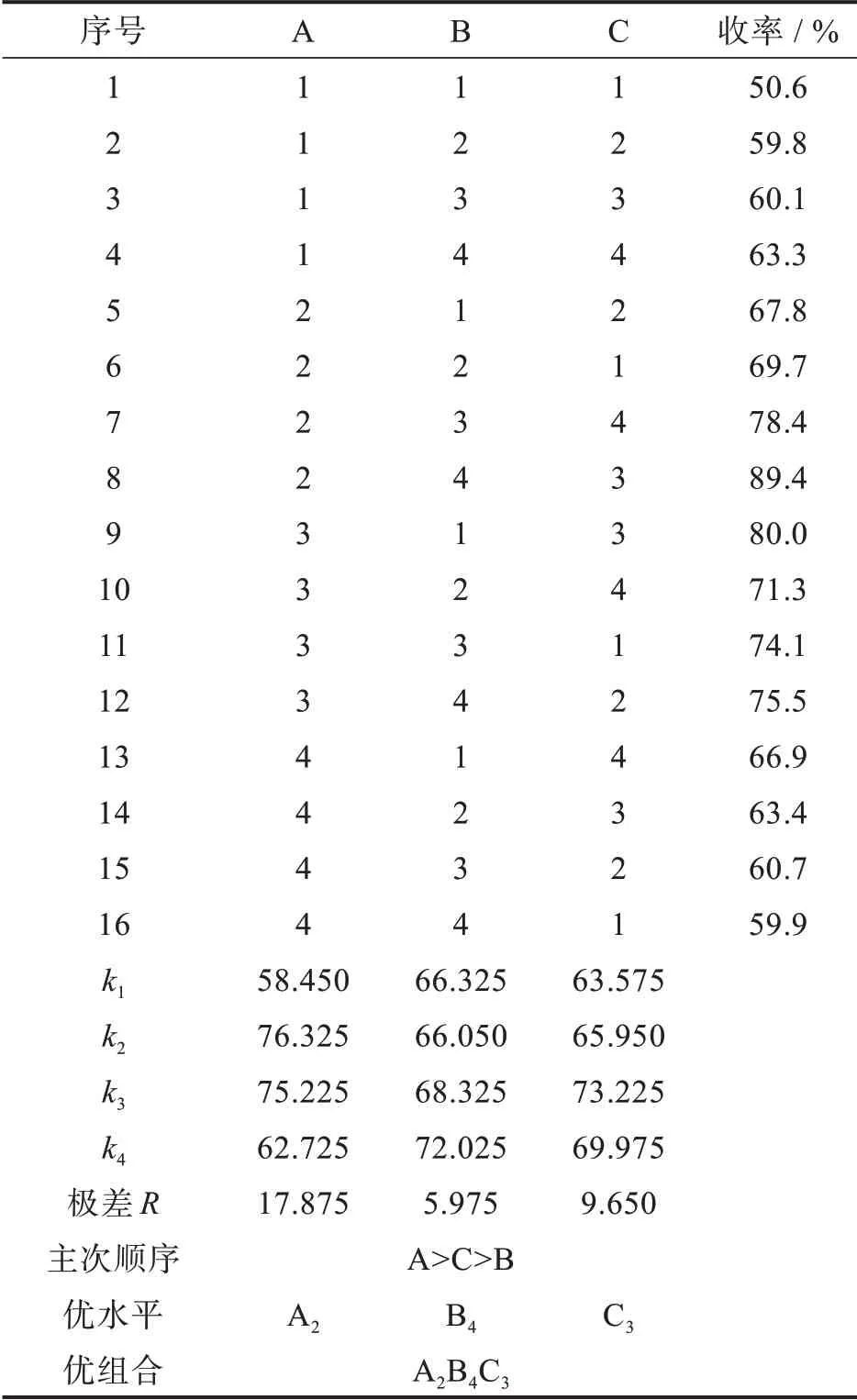
表4 氨基化反应正交实验结果Tab.4 Orthogonal experimental results of amination reaction
从计算出的K值得出A2、B4、C3最佳。通过公式计算出本实验中各Rj的值得到:R1>R3>R2,表明各因子A、C、B对氨基化实验的影响逐渐减小。因此合成化合物Ⅲ的最优工艺条件为:A2B4C3,即n(Ⅱ)∶n(4-异丙氨基丁醇)=1.0∶4.0,反应在170℃进行68 h。过量的4-异丙氨基丁醇减压浓缩回收,避免了其它合成方法后处理时大量废水的产生。
2.4 2-{(4-[N-(5,6-二苯基吡嗪-2-基)-N-异丙基氨基]丁氧基}乙酸甲酯(Ⅳ)的合成
化合物Ⅲ与溴乙酸甲酯在氢氧化钠为缚酸剂、四丁基溴化铵为相转移催化剂条件下,发生Williamson反应生成化合物Ⅳ。物料比、溶剂、反应温度和时间对反应的影响见表5。当n(Ⅲ)∶n(溴乙酸甲酯)∶n(氢氧化钠)∶n(四丁基溴化铵)=1.0∶1.0∶15.0∶0.1时,以二氯甲烷为溶剂,由于回流温度低,化合物Ⅲ未完全转化为Ⅳ,化合物Ⅳ的收率约为75%(entry 1,2);以环己烷为溶剂回流,化合物Ⅲ也未完全转化为Ⅳ;以甲苯为溶剂80℃反应5 h,化合物Ⅲ完全转化,Ⅳ的收率达到96.2%(entry 6)。升高甲苯为溶剂时的反应温度至90℃以上,虽然化合物Ⅲ完全转化,但Ⅳ的收率却降低,原因是高温会发生酯基水解。降低物料比中氢氧化钠的量,即使延长反应时间,化合物Ⅲ也未完全转化,且Ⅳ的收率却降低。因此,化合物Ⅲ与溴乙酸甲酯在碱性条件下反应生成Ⅳ的最佳条件为:n(Ⅲ)∶n(溴乙酸甲酯)∶n(氢氧化钠)∶n(四丁基溴化铵)=1.0∶1.0∶15.0∶0.1,甲苯为溶剂,80℃反应5 h。
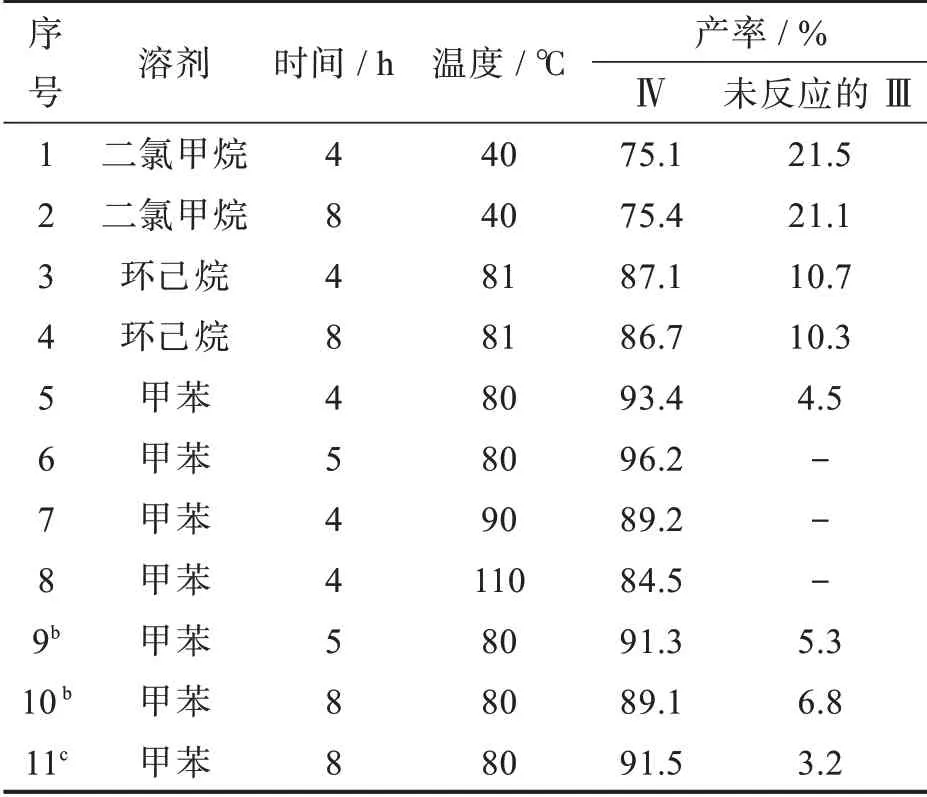
表5 不同反应条件对合成化合物Ⅳ的影响aTab.5 Effect of different reaction conditions on formation of compoundⅣa
2.5 Selexipag的合成
为了将化合物Ⅳ转化为酰胺化产物,浅木哲夫等[11]先将化合物Ⅳ水解成酸,再在CDI、TBTU和DCC存在下与甲基磺酰胺反应得到Selexipag,操作繁琐,杂质多。最近,Zhang等[19]将不活泼酯、胺和叔丁醇钠混合,在不加溶剂条件下室温反应直接得到系列酰胺化产物,为酯的酰胺化反应开辟了一条绿色合成路线。本研究将化合物Ⅳ与甲基磺酰胺在叔丁醇钠存在下室温反应同样得到了酰胺化产物Selexipag。化合物Ⅳ、甲基磺酰胺和叔丁醇钠摩尔比以及反应时间对酰胺化反应的影响见表6。当n(甲基磺酰胺)∶n(Ⅳ)∶n(叔丁醇钠)为1.0∶2.0∶1.5时,室温下反应1 h,Selexipag的收率为85.8%(entry 1)。减少n(Ⅳ)的投料,Selexipag的收率逐渐下降,其中n(甲基磺酰胺)∶n(Ⅳ)∶n(叔丁醇钠)为1.0∶1.05∶1.5时,Selexipag的收率为80.1%(entry 5),将反应时间延长到2 h,Selexipag的收率达到最高值92.3%(entry 9),进一步延长反应时间,收率基本不变;减少叔丁醇钠的用量,Selexipag的收率降低(entry 11,12)。当甲基磺酰胺相对化合物Ⅳ过量时,Selexipag的收率降低至57.2%(entry 7)。因此,化合物Ⅳ与甲基磺酰胺发生酰胺化反应的最佳条件为n(甲基磺酰胺)∶n(Ⅳ)∶n(叔丁醇钠)为1.0∶1.05∶1.5,室温下反应2 h。反应后处理阶段,未反应完的酯皂化溶于水中除掉,操作简单。
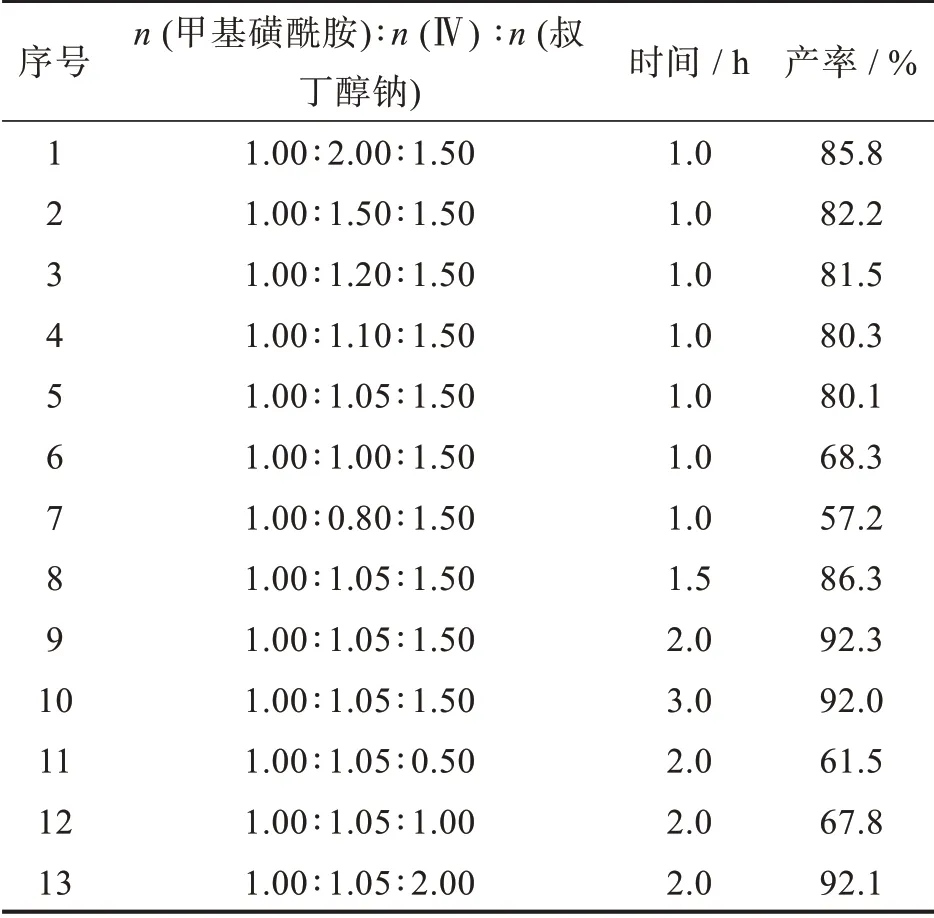
表6 化合物Ⅳ酰胺化条件的优化Tab.6 Optimization of amidation conditions of compoundⅣ
3 结 论
以二苯基乙二酮和氨基乙酰胺为起始物料,通过环合反应合成了5,6-二苯基吡嗪-2-醇。在无缚酸剂和减少三氯氧磷用量条件下,以DMF为溶剂,将5,6-二苯基吡嗪-2-醇的羟基氯代得到2-氯-5,6-二苯基吡嗪。2-氯-5,6-二苯基吡嗪与适量4-异丙氨基丁醇反应,得到4-[N-(5,6-二苯基-2-基)(N-异丙基)氨基]-1-丁醇,4-异丙氨基丁醇既是反应试剂,又是溶剂和缚酸剂,过量的4-异丙氨基丁醇回收利用,避免了加入其它加缚酸剂和溶剂后处理时大量废物的产生。以氢氧化钠为碱、四丁基溴化铵为相转移催化剂,4-[N-(5,6-二苯基-2-基)(N-异丙基)氨基]-1-丁醇与溴乙酸甲酯在甲苯中反应,生成2-{4-[N-(5,6-二苯基吡嗪-2-基)-N-异丙基氨基]丁氧基}乙酸甲酯。后者和甲基磺酰胺在叔丁醇钠作用下直接发生酰胺化反应生成Selexipag,该反应无需溶剂,室温反应,后处理简单。优化后的合成路线Selexipag的收率达到74.5%,合成步骤少,反应条件温和,产物易于分离纯化,无需特殊设备,适合工业化生产。
