1-脱氧野尻霉素的合成工艺研究
2022-02-28王峰峰方志杰
王峰峰,方志杰
(南京理工大学化学与化工学院,江苏 南京 210014)
目前,临床用于治疗糖尿病的药物大部分存在着较为严重的不良反应。而α-葡萄糖苷酶抑制剂作为20世纪新兴的一类治疗糖尿病的药物,具有用量少、作用显著等特点,其中1-脱氧野尻霉素(1-deoxynojirimycin,DNJ)是国际公认的唯一零伤害的生物制剂,其化学名为(2R,3R,4R,5S)-2-羟甲基哌啶-3,4,5-三醇。目前,全球仅有30家左右的DNJ供应商,其中包含了相当一部分经销商,这些供应商绝大多数来自中国,其中60%~70%的供应商仅能提供试剂级的小样或标准品,能提供大批量DNJ的很少。随着DNJ开发力度的加大,近年来国内外市场对DNJ的需求不断增加,但DNJ的合成研究和产能明显落后,使其推广应用受到限制。因此,对DNJ产业化合成路线的研究受到研究者的广泛重视。
通过对DNJ的结构分析,发现其手性中心在2,3,4,5位,与葡萄糖的结构较为相似,因此以葡萄糖或葡萄糖衍生物合成DNJ不仅可以大大降低DNJ合成难度,而且从原子经济学的角度来说无疑是最合适的方法。基于相关文献[1-13],作者以商业化的2,3,4,6-O-四苄基-D-吡喃葡萄糖(SM)为起始原料,经羟基氧化、内酯氨解后开环得到(2R,3S,4R,5R)-2,3,4,6-四(苄氧基)-5-羟基己酰胺(D-1);化合物D-1通过斯文氧化得到粗产物,再通过正庚烷-乙酸乙酯纯化得到(2R,3S,4S)-2,3,4,6-四(苄氧基)-5-氧代六酰胺(D-2);化合物D-2经氰基硼氢化钠还原胺化后再次关环,然后用甲醇精制得到(3R,4S,5R,6R)-3,4,5-三(苄氧基)-6((苄氧基)甲基)哌啶-2-酮(D-3);化合物D-3经硼氢化钠和三氟化硼原位还原得到油状粗产物,然后在乙酸乙酯中酸化成盐得到2,3,4,6-四苄基-1-脱氧野尻霉素盐酸盐(D-4);化合物D-4经Pd(OH)2/C加氢脱苄基反应,最后在甲醇中析晶得到目标化合物DNJ;并对中间体和目标化合物的结构进行确认。该合成路线共5步反应,如图1所示。
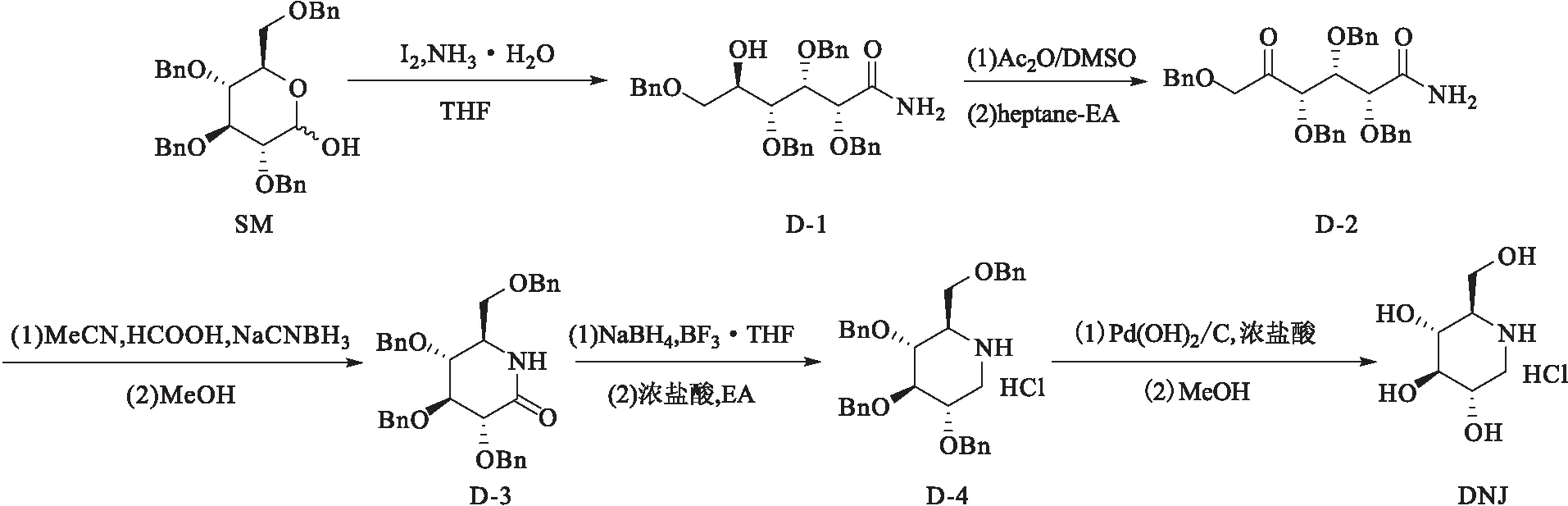
图1 DNJ的合成路线
1 实验
1.1 试剂与仪器
2,3,4,6-O-四苄基-D-吡喃葡萄糖,上海皓鸿生物医药科技有限公司;四氢呋喃(THF)、氨水、二甲基亚砜(DMSO)、醋酐(Ac2O)、无水硫酸钠、乙腈、甲酸、碳酸氢钠、正庚烷、三氟化硼四氢呋喃络合物(BF3·THF)、硼氢化钠、浓盐酸、甲醇等均为分析纯,国药集团化学试剂有限公司;碘,分析纯,江苏艾康生物医药研发有限公司;氰基硼氢化钠,分析纯,上海迈瑞尔化学技术有限公司;20%Pd(OH)2/C,陕西瑞科新材料股份有限公司;乙酸乙酯(EA)、二氯甲烷(DCM),上海佳足实业有限公司。
SHZ-D(Ⅲ)型循环水式多用真空泵、RE-2000型旋转蒸发器、98-2型磁力搅拌器,上海积坤实验仪器有限公司;JA2003型电子天平,上海舜宇恒平科学仪器有限公司;DFY-5L/25型低温恒温浴、DZF-6050型真空干燥箱,卯氏(上海)实验仪器有限公司;HWCL-3型集热式恒温磁力搅拌浴,郑州长城科工贸有限公司。
1.2 方法
1.2.1 化合物D-1的合成
向21.4 g(39.58 mmol)2,3,4,6-O-四苄基-D-吡喃葡萄糖(SM)中加入43 mL(2 V,V表示以原料计的液料比,mL∶g,下同)THF,搅拌均匀,然后依次加入12.0 g(47.28 mmol,1.2 eq.)碘、118 mL(5.5 V)氨水(25%~28%),密闭条件下于10~20 ℃保温反应20 h;反应结束后加入4.8 g五水硫代硫酸钠搅拌20 min,反应液于30~35 ℃减压浓缩至馏分明显减少;降温至10~25 ℃,加入100 mL×2二氯甲烷萃取2次,合并二氯甲烷层;用30 mL食盐水(15%)洗涤1次,分液;取二氯甲烷层,减压旋干得到23.2 g淡黄色糖浆物(D-1),将其溶于60 mL DMSO中,待用。
1.2.2 化合物D-2的合成
室温下将86 mL Ac2O滴加到86 mL DMSO中配制Ac2O/DMSO溶液,降温至0~5 ℃,控温0~10 ℃将化合物D-1的DMSO溶液滴加到Ac2O/DMSO溶液中,维持内温5~10 ℃(最佳6~8 ℃)保温反应20 h;控温10~15 ℃向反应液中滴加540 mL水,搅拌15 min,依次用200 mL和100 mL二氯甲烷萃取2次,合并二氯甲烷层;加入300 mL水,缓慢加入50~60 g碳酸钠调节水层pH值为8~9,分液;取二氯甲烷层,加入100 mL水洗涤1次;取二氯甲烷层,减压旋干得到23.6 g黄色糖浆物(D-2粗品);加入44 mL乙酸乙酯,将D-2粗品搅拌溶解,控温15~25 ℃缓慢滴加176 mL正庚烷搅拌析晶2 h;抽滤,滤饼加入15 mL正庚烷-乙酸乙酯(4∶1,体积比,下同)淋洗;45~50 ℃减压干燥得到14.58 g类白色固体(D-2),收率66.56%(SM到D-2的两步收率)。m.p.113.6~116.0 ℃;1HNMR(DMSO,400 MHz),δ:7.53(s, 1H),7.43(s,1H),7.40~7.21(m,20H),4.69~4.51(m,5H),4.43~4.34(m,5H),4.31(d,J=4.1 Hz,1H),4.15(t,J=4.7 Hz,1H),4.03(d,J=5.1 Hz,1H)。
1.2.3 化合物D-3的合成
取12.0 g 化合物D-2,加入120 mL(10 V)乙腈、24 mL(2 V)甲酸、3.42 g(2.5 eq.)氰基硼氢化钠,升温至80~85 ℃反应3 h;降至室温,过滤,滤饼用12 mL乙腈淋洗,合并滤液于45 ℃减压旋干回收溶剂;向旋干物加入50 mL二氯甲烷、50 mL水,搅拌分液;取二氯甲烷层用30 mL饱和碳酸氢钠溶液洗涤1次,分液;取二氯甲烷层再用30 mL食盐水洗涤1次,无水硫酸钠干燥,减压旋干得到10.88 g黄色固体(D-3粗品);加入33 mL(3 V)甲醇升温至回流溶清,降温至0~5 ℃搅拌析晶3 h;抽滤,滤饼用3 mL冰甲醇淋洗,45~50 ℃减压干燥得到7.62 g白色固体(D-3),收率65.4%。1HNMR(CDCl3,500 MHz),δ:3.26(t,J=8 Hz,1H),3.28~3.43(m,1H),3.43~3.63(m,2H),3.92(t,J=8.1 Hz,1H),4.01(d,J=8 Hz,1H),4.46~4.51(m,3H),4.73~4.79(m,2H),4.84~4.88(m,2H),5.19(d,J=11.2 Hz,1H),5.95(s,1H),7.19~7.44(m,20H);13CNMR(CDCl3,125 MHz),δ:169.86,137.11,136.92,136.67,136.04,127.62,127.46,127.36,127.22,127.11,127.09,127.00,126.97,126.89,81.35,77.84,76.46,76.40,76.20,76.08,75.95,73.81,73.71,72.40,68.87,52.99。
1.2.4 化合物D-4的合成
取6.5 g 化合物D-3,加入65 mL(10 V)THF溶解;氮气保护下加入1.4 g(3.0 eq.)硼氢化钠,降温至0 ℃左右;控温0~5 ℃滴加6.77 g(4.0 eq.)BF3·THF溶液,回温至15~20 ℃保温反应2 h;缓慢滴加16.3 mL甲醇淬灭反应,滴加6.5 mL浓盐酸,15~20 ℃保温反应16 h;加入32.5 mL水搅拌均匀,滴加约78 mL碳酸钠溶液(20%)调节反应液pH值至9~10,加入65 mL×2乙酸乙酯萃取2次,合并乙酸乙酯层;用65 mL饱和食盐水洗涤1次,再用无水硫酸钠干燥,45~50 ℃减压浓缩至反应液体积约为70~80 mL;缓慢滴加4 mL含氯化氢的乙酸乙酯溶液(4 mol·L-1),降温至15~20 ℃强力搅拌析晶3 h;过滤,45~50 ℃减压干燥得到5.52 g类白色固体(D-4),收率81.5%。1HNMR(CDCl3,500 MHz),δ:2.54(dd,J=8 Hz、12 Hz,1H),2.77(ddd,J=3 Hz、6 Hz、9 Hz,1H),3.28(dd,J=4.5 Hz、12 Hz,1H),3.40(t,J=9 Hz,1H),3.51~3.61(m,3H),3.70(dd,J=3 Hz、9 Hz,1H),4.46~4.54(m,3H),4.69~4.76(m,2H),4.87~4.92(m,2H),5.02(d,1H),7.23~7.40(m,20H);13CNMR(CDCl3,125 MHz),δ:137.98,137.56,137.46,137.04,127.47,127.08,127.01,126.94,126.85,126.75,126.59,86.38,79.69,79.13,76.36,76.10,75.85,74.74,74.26,72.47,71.86,69.29,58.80,47.16。
1.2.5 目标化合物DNJ的合成
取3.0 g 化合物D-4,加入120 mL甲醇搅拌溶解;氮气球置换反应瓶内气体3次,加入2 mL浓盐酸、450 mg 20%Pd(OH)2/C;氮气球再次置换反应瓶内气体3次,然后氢气球置换反应瓶内气体3次,15~25 ℃保温反应约48 h;过滤,滤饼用20 mL纯化水淋洗,将滤液减压浓缩得到1.107 g油状物(DNJ粗品);加入4 mL甲醇升温至回流保温0.5 h,缓慢降温至0~5 ℃析晶3 h;过滤,滤饼用1 mL甲醇淋洗,50~55 ℃减压干燥得到0.9 g白色固体(DNJ),收率84.2%。1HNMR(D2O,500 MHz),δ:3.73(dd,J=11.6 Hz、2.8 Hz, 1H),3.53(dd,J=11.7 Hz、6.3 Hz,1H),3.44~3.34(m,1H),3.22(t,J=9.1 Hz,1H),3.13(t,J=9.5 Hz,1H),3.02(dd,J=12.3 Hz、5.1 Hz,1H),2.50~2.41(m,1H),2.36(t,J=11.6 Hz,1H);13CNMR(D2O,125 MHz),δ:77.35,70.47,69.85,60.33,59.47,47.66。
2 结果与讨论
2.1 化合物D-1的合成工艺优化
实验发现,SM合成D-1的过程副反应很少,产物D-1中几乎没有杂质,因此以原料的转化情况作为评价标准,考察反应物(THF、氨水、碘)投料量对化合物D-1合成的影响,结果见表1。
由表1可知:(1)在THF投料量为4.2 V、氨水投料量为14.5 V时,将碘投料量从1.3 eq.降至1.2 eq.时原料均能反应完全,继续降至1.1 eq.时有少量原料残留,因此,确定最佳碘投料量为1.2 eq.。(2)在THF投料量为4.2 V、碘投料量为1.2 eq.时,将氨水投料量从14.5 V分别降至10.0 V、5.5 V时原料均能反应完全,因此,确定最佳氨水投料量为5.5 V。(3)在氨水投料量为5.5 V、碘投料量为1.2 eq.时,将THF投料量从4.2 V分别降至3 V、2 V时原料均能反应完全,继续降至1 V时有少量原料残留,因此,确定最佳THF投料量为2 V。
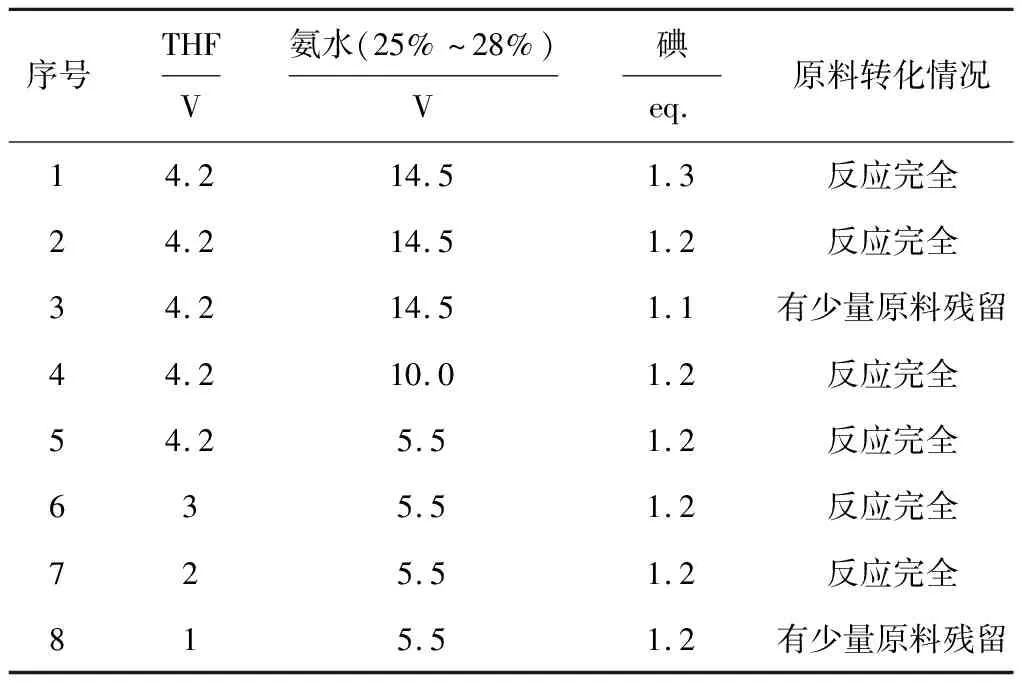
表1 反应物投料量对化合物D-1合成的影响
2.2 化合物D-2的合成工艺优化
2.2.1 反应温度的优化
实验发现,由化合物D-1合成化合物D-2的过程对温度较为敏感。在不同温度下,按1.2.2方法合成化合物D-2,考察反应温度对化合物D-2合成的影响,结果见表2。

表2 反应温度对化合物D-2合成的影响
由表2可知,随着反应温度的降低,化合物D-2的收率逐渐升高。当反应温度为15~20 ℃时,化合物D-2的收率为48.0%;当反应温度降至5~10 ℃时,收率提高到65.2%;继续降低反应温度至0 ℃,反应无法正常进行。因此,确定最佳反应温度为5~10 ℃。
2.2.2 纯化工艺优化
由于下步合成化合物D-3时用到的氰基硼氢化钠价格较为昂贵,而且上步产物D-1没有提纯,因此化合物D-2中会残留一些杂质。对化合物D-2进行纯化可以提高D-2的有效含量,这样不仅可以减少下步反应氰基硼氢化钠的用量,而且可以减少副产物的生成,减轻后续纯化的压力。基于此,采用正庚烷-乙酸乙酯对化合物D-2进行纯化,考察正庚烷与乙酸乙酯的体积比(V正庚烷∶V乙酸乙酯)对纯化收率的影响,结果见表3。
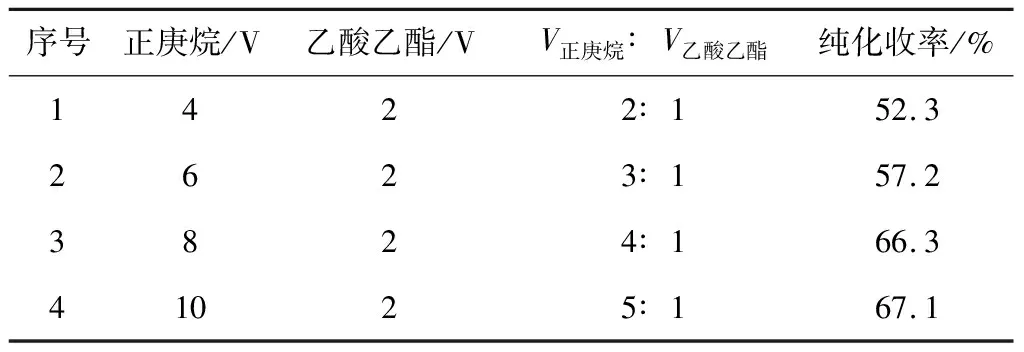
表3 正庚烷与乙酸乙酯的体积比对化合物D-2纯化收率的影响
由表3可知,固定乙酸乙酯投料量为2 V,逐渐增加正庚烷投料量,化合物D-2的纯化收率随着V正庚烷∶V乙酸乙酯的增大逐渐升高;当V正庚烷∶V乙酸乙酯为2∶1时,纯化收率为52.3%;当V正庚烷∶V乙酸乙酯为4∶1时,纯化收率提高到66.3%;当V正庚烷∶V乙酸乙酯继续增大至5∶1时,纯化收率升幅不明显,仅为67.1%。因此,确定最佳V正庚烷∶V乙酸乙酯为4∶1。
2.3 化合物D-3的合成工艺优化
2.3.1 反应物投料量的优化
以化合物D-2为原料合成化合物D-3,考察反应物(乙腈、甲酸、氰基硼氢化钠)投料量对化合物D-3合成的影响,结果见表4。
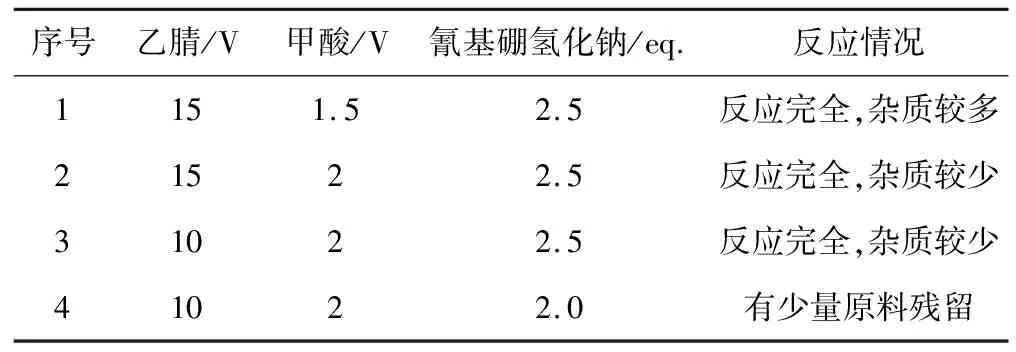
表4 反应物投料量对化合物D-3合成的影响
由表4可知:(1)当乙腈投料量为15 V、氰基硼氢化钠投料量为2.5 eq.时,将甲酸投料量从1.5 V增至2 V时,杂质明显减少,因此,确定最佳甲酸投料量为2 V。(2)当甲酸投料量为2 V、氰基硼氢化钠投料量为2.5 eq.时,将乙腈投料量从15 V减至10 V时,对反应影响不大,因此,确定最佳乙腈投料量为10 V。(3)当乙腈投料量为10 V、甲酸投料量为2 V时,将氰基硼氢化钠投料量从2.5 eq.减至2.0 eq.时,会有少量原料残留,因此,确定最佳氰基硼氢化钠投料量为2.5 eq.。
2.3.2 纯化工艺优化
化合物D-2经过还原反应得到5位R型D-3的同时,会有少量的5位S型异构体杂质产生。基于5位R型D-3为固体、5位S型异构体杂质为油状物,可通过纯化去除异构体杂质。分别采用正庚烷-乙酸乙酯、甲醇对化合物D-3进行纯化,考察纯化溶剂及其用量对化合物D-3纯化收率的影响,结果见表5。

表5 纯化溶剂及其用量对化合物D-3纯化收率的影响
由表5可知,以正庚烷-乙酸乙酯(3∶1)作为纯化溶剂时,用量需要20 V,D-3纯化收率为62.4%;以正庚烷-乙酸乙酯(4∶1)作为纯化溶剂时,用量需要25 V,纯化收率为67.2%;以甲醇作为纯化溶剂时,用量只需要3 V,纯化收率达到71.5%。以甲醇作为纯化溶剂时不仅用量少而且纯化收率高,因此,选择甲醇作为纯化溶剂纯化D-3粗品。
2.4 化合物D-4的合成工艺优化
2.4.1 还原剂的优化
以化合物D-3为原料,通过硼氢化钠和三氟化硼原位还原合成化合物D-4。考察不同还原剂对化合物D-4合成的影响,结果见表6。

表6 不同还原剂对化合物D-4合成的影响
由表6可知,分别以NaBH4+BF3·Et2O、NaBH4+BF3·THF为还原剂时,原料均能反应完全,而且杂质很少;以NaBH4+BF3·ACN为还原剂时,原料虽能反应完全,但杂质较多。由于该步反应溶剂为THF,因此,确定最佳还原剂为NaBH4+BF3·THF。
2.4.2 还原剂投料比的优化
还原剂NaBH4+BF3·THF的反应原理为:NaBH4与BF3反应生成硼烷,硼烷通过原位还原生成内酰胺。在确定化合物D-4合成反应的还原剂为NaBH4+BF3·THF后,对NaBH4与BF3·THF的投料比进行优化,结果见表7。
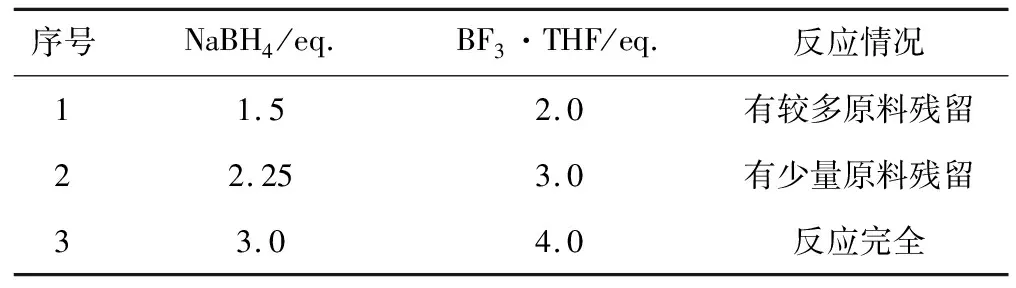
表7 NaBH4与BF3·THF的投料比对化合物D-4合成的影响
由表7可知,当NaBH4投料量为1.5 eq.、BF3·THF投料量为2.0 eq.时,有较多原料未反应完;提高两者投料量分别至2.25 eq.、3.0 eq.时,有少量原料未反应完;继续提高两者投料量分别至3.0 eq.、4.0 eq.时,原料反应完全。因此,确定最佳NaBH4投料量为3.0 eq.、BF3·THF投料量为4.0 eq.。
2.4.3 硼烷胺络合物解离时间的优化
使用NaBH4+BF3·THF原位还原酰胺的羰基为亚甲基后,并不能直接生成所需的产物,而是生成了硼烷胺络合物,需要加浓盐酸使其完全解离为游离胺,因此需要对解离时间进行优化,结果见表8。

表8 硼烷胺络合物解离时间的优化
由表8可知,随着解离时间的延长,硼烷胺络合物的转化率逐渐升高;当解离时间为2 h时,约15%的硼烷胺络合物转化为游离胺,转化率较低;当解离时间延长至16 h时,硼烷胺络合物转化完全。因此,确定最佳解离时间为16 h。
2.4.4 成盐溶剂的优化
原料的纯度直接影响目标化合物DNJ的纯度。由于化合物D-3原位还原得到的粗产物是油状物,纯化起来较为麻烦,但是它的盐酸盐(化合物D-4)是固体,可以在某些有机溶剂中通过沉淀的方式分离出来,因而可以采用成盐溶剂对油状粗产物进行纯化。通过对常规溶剂进行筛选,发现化合物D-4在二氯甲烷或氯仿中极易溶解,而在醇类溶剂中溶解度较大,因此,选择甲叔醚、丙酮、乙酸乙酯等3种溶剂对油状粗产物进行纯化,考察成盐溶剂对化合物D-4纯化收率的影响,结果见表9。
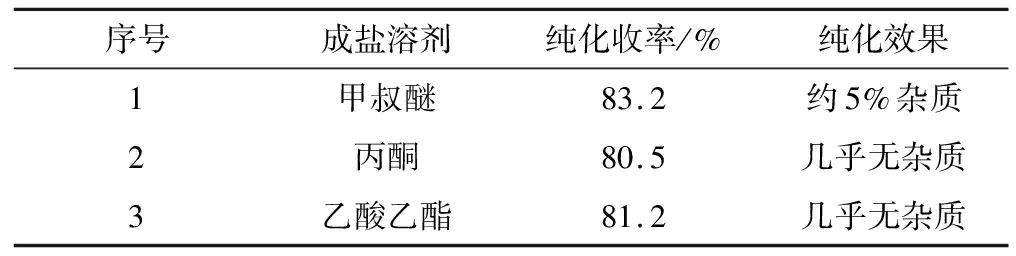
表9 成盐溶剂对化合物D-4纯化收率的影响
由表9可知,以甲叔醚为成盐溶剂时,化合物D-4的纯化收率最高,为83.2%,但含有约5%的杂质;以丙酮或乙酸乙酯为成盐溶剂时,纯化收率相当,几乎无杂质,纯化效果均较好。考虑到反应后处理的萃取溶剂是乙酸乙酯,实际操作时只需要浓缩出部分乙酸乙酯,即可进行酸化成盐析晶,操作简便。因此,确定乙酸乙酯为成盐溶剂。
2.5 目标化合物DNJ的合成工艺优化
以化合物D-4为原料,通过脱苄基反应合成目标化合物DNJ。考察不同脱苄基试剂组合对DNJ合成的影响,结果见表10。

表10 不同脱苄基试剂组合对目标化合物DNJ合成的影响
由表10可知:(1)在以10%Pd/C为催化剂的情况下,以甲酸为氢源时的DNJ收率约10%,以甲酸铵为氢源时的DNJ收率约15%,以氢气为氢源时的DNJ收率约85%。因此,确定最佳氢源为氢气。(2)将催化剂10%Pd/C替换为20%Pd(OH)2/C,DNJ收率提升到95%以上,而且中间态很少。因此,确定最佳催化剂为20%Pd(OH)2/C。
3 结论
以2,3,4,6-O-四苄基-D-吡喃葡萄糖为起始原料(SM),经羟基氧化、内酯氨解后得到(2R,3S,4R,5R)-2,3,4,6-四(苄氧基)-5-羟基己酰胺(D-1),物料比m(SM)∶V(THF)∶V(氨水)为1∶2∶5.5,氧化剂用量n(SM)∶n(碘)为1∶1.2;化合物D-1通过斯文氧化、正庚烷-乙酸乙酯纯化后得到(2R,3S,4S)-2,3,4,6-四(苄氧基)-5-氧代六酰胺(D-2),反应温度为5~10 ℃,纯化工艺投料比m(D-2粗品)∶V(乙酸乙酯)∶V(正庚烷)为1∶2∶8;化合物D-2经氰基硼氢化钠还原胺化、甲醇纯化后得到(3R,4S,5R,6R)-3,4,5-三(苄氧基)-6((苄氧基)甲基)哌啶-2-酮(D-3),物料比m(D-2)∶V(乙腈)∶V(甲酸)为1∶10∶2,还原剂用量n(D-2)∶n(氰基硼氢化钠)为1∶2.5,纯化溶剂甲醇用量m(D-3粗品)∶V(甲醇)为1∶3;化合物D-3在四氢呋喃中经硼氢化钠和三氟化硼原位还原得到油状粗产物,然后在乙酸乙酯中酸化成盐得到2,3,4,6-四苄基-1-脱氧野尻霉素盐酸盐(D-4),物料比n(D-3)∶n(NaBH4)∶n(BF3·THF)为1∶3.0∶4.0,后处理时硼烷胺络合物最佳解离时间为16 h,成盐溶剂优选乙酸乙酯;最后化合物D-4经Pd(OH)2/C加氢、并在浓盐酸的催化下通过脱苄基反应得到1-脱氧野尻霉素(DNJ)。总计5步反应,其中从SM到D-2的两步收率为66.56%,D-2到D-3的收率为65.4%,D-3到D-4的收率为81.5%,D-4到DNJ的收率为84.2%,总收率在30%左右。该方法操作简便、收率较高,每一步所用到的化学试剂以及实验操作在放大批量时均是可行的,可满足大规模生产的要求。
