视网膜色素变性患者RP1基因突变位点与临床表型相关性初探
2022-02-11崔福玲孟令强马志国
崔福玲,孟令强,王 静,马志国
1.山东省滨州市人民医院康复医学科,山东滨州,256610; 2.山东省东营市中医院检验科,山东东营 257000;山东省德州市人民医院:3.眼科;4.检验科,山东德州 253000
视网膜色素变性是视网膜变性里最常见的一组以进行性光感受器细胞及视网膜色素上皮功能丧失为共同表现的致盲性眼病,在临床表型和潜在的遗传缺陷方面具有很强的异质性[1]。RP发病率为1/3 500~1/5 000,影响全球约150万人[2]。近年来视网膜色素变性基因的发现主要依靠二代测序技术,二代测序技术能够并行测序所有已知的相关基因,从预选的基因组区域产生数百万个读数[3]。有研究发现,所有突变患者发病年龄均在21岁以下,提示可能没有必要对50岁以上的患者进行常规筛查[4]。本研究对本院临床确诊为视网膜色素变性的人群进行二代测序技术测序和Sanger验证,分析总结视网膜色素变性的视网膜色素变性1基因(RP1基因)突变及其引起的特殊临床表现特征,为后续临床确诊和遗传咨询等工作提供帮助。
1 资料与方法
1.1一般资料 本研究采用横断面研究设计。将2018年1月至2019年7月在山东省滨州市人民医院确诊的视网膜色素变性25个家系共86例患者纳入本研究。纳入标准:(1)夜盲史;(2)视力逐渐下降;(3)典型眼底改变,视盘呈蜡黄色萎缩,视网膜有骨细胞样色素沉着,血管变细,视网膜呈青灰色;(4)早期周边视野呈环形暗点,晚期视野呈向中心缩窄;(5)病变早期即可出现视网膜电图(ERG)、眼电图(EOG)明显异常。其中,男43例,女43例;年龄9~88岁,平均(49.44±3.67)岁。排除标准:(1)不能配合完成相关检查者;(2)无法判断发病原因是遗传因素引起者;(3)无家系血样可以进行基因验证者。
患者和家系成员均通过标准临床检查,以明确诊断,并排除其他非遗传因素引起的眼部疾病。临床检查包括病史询问、物理检查等。其中,病史询问包括基础个人信息(包括性别、年龄、籍贯、民族等),主诉,现病史(初次发病时间、发病规律、就诊情况、用药情况、并发症种类和时间等),既往史(全身情况、基因检测史等),家族遗传史,手术和药物使用史,婚育史等。物理检查包括视力和矫正视力、非接触性眼压、色觉、B超(天津迈达,型号ODM-2200)、视野(卡尔蔡司,型号750i)、眼前节照相、眼底彩色照相(Topcon,型号TRC NW-300)、光相干断层扫描(OCT,型号Cirrus HD-OCT 5000)。除此之外,患者还进行常规肘部静脉采血,采用高通量二代基因测序技术进行基因测序[5]。
1.2样本采集 将符合要求的患者及家系成员常规采集肘部静脉血5 mL,ETDA抗凝,-80 ℃保存。
1.3提取基因组DNA
1.3.1DNA提取 QIAGEN试剂盒提取外周血的DNA,Qubit 荧光仪检测DNA浓度,琼脂糖凝胶电泳检测DNA完整性。
1.3.2文库制备 参考华大基因的BGISEQ-500的文库构建指导手册进行。(1)将质量合格的DNA样品通过超声波高性能样品处理系统(Covaris)随机打断,经过片段选择后得到150~250 bp的片段;(2)随后进行DNA片段末端修复,3′端加上“A”碱基,两端加上文库接头;(3)接头连接后的文库进行线性扩增(LM-PCR)制备成杂交文库;(4)杂交文库与眼科基因芯片(深圳华大生命科学研究院提供792个基因Panel,其中视网膜色素变性基因78个)进行捕获富集,洗脱未富集的片段后进行扩增;(5)扩增产物进行单链分离和环化处理,环化文库进行滚环复制生成DNA纳米球。采用Qubit荧光仪检测文库浓度进行质量控制。
1.3.3上机测序 质量控制合格的文库采用BGISEQ-500 PE50+10程序上机测序,共检测792个眼科目标基因,其中含有78个视网膜色素变性相关基因。
1.3.4信息分析 (1)从测序仪获取原始数据(FASTQ数据);(2)过滤:对原始FASTQ数据进行质量控制,去除低质量数据;(3)比对:利用SOAP和BWA软件,使用hg19参考序列进行比对;(4)去重复:基于Picard的去重复算法,去除重复序列;(5)变异检测:基于GATK对数据进行变异检测;(6)变异注释:使用Next GENe5.4.5分析软件对基因变异与特殊临床表型的相关性进行分析;使用频率数据库dbSNP、千人基因组数据库、ESP6500数据库、ExAC数据库及BGI内部数据库进行频率注释;使用HGVS对变异进行标准命名;使用OMIM、HGMD等疾病数据库进行突变及疾病注释。
1.4统计学处理 采用SPSS20.0统计学软件进行数据处理及统计分析。基因突变以百分率表示,组间比较采用χ2检验。P<0.05为差异有统计学意义。
2 结 果
2.1测序结果 25个家系共86例患者,目标序列平均测序深度为107.79,目标序列覆盖率平均99.93%。平均99.98%目标序列覆盖率>10×,平均95.41%的目标序列覆盖率>30×,测序深度和平均覆盖率均符合检测要求。
2.2基因检出情况 25个家系先证者中,检测出已知致病性基因突变病例13例;检出疑似致病性基因突变病例9例;临床意义未明基因突变有3例;未检出阴性病例,25个家系均有阳性发现。8号患者的RP1基因上有2个突变位点,分别为c.2886delA和c.4129delG突变。25号患者的RP1基因上有4个致病突变位点,分别为c.4168_4169insT、c.4196delG、c.4169A>G和c.6353G>A突变。在检出的基因变异位点中,RP1基因发现9个未报道致病基因突变位点,见表1。
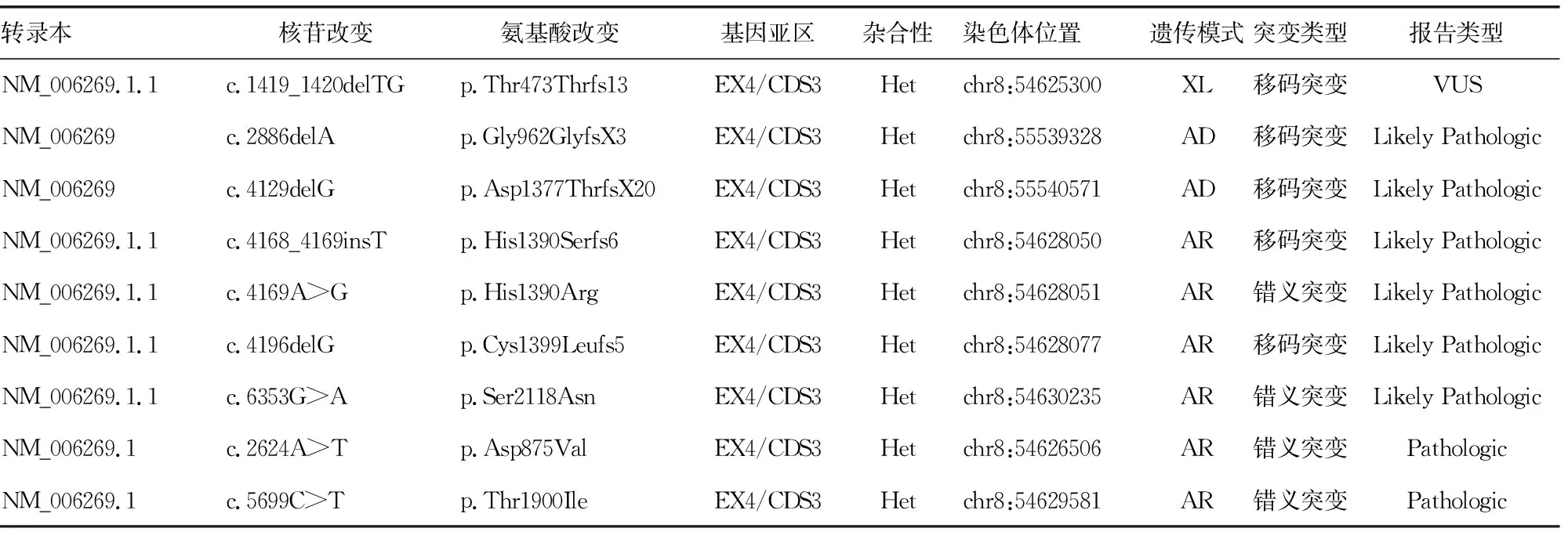
表1 视网膜色素变性家系基因检测中发现未报道的RP1基因位点突变
2.3临床观察 8号病例:眼视盘界清色蜡黄,血管纤细,视网膜萎缩,呈青灰色,后极黄斑旁及赤道部少量骨细胞样色素析出,眼底照相提示,色素散在分布,未附着于血管旁。自发荧光照相提示,视网膜色素背景荧光变淡,左眼黄斑旁机化瘢痕处点状强荧光。视野检查提示,双眼视野呈不规则片状,左眼中央区视野尚存。见图1。25号病例:眼底照相显示,双眼视盘界清色淡,血管极细,黄斑中心凹处色暗,除后极黄斑区外,其余视网膜呈青灰色,未见明显骨细胞样色素沉着。自发荧光照相提示,眼底色素背景荧光极低或消失,呈斑驳样。双眼视野检查提示,视野呈管状,周边视野完全丧失。OCT提示双眼黄斑中心凹处极度萎缩,厚度仅为右眼37 μm,左眼31 μm。椭圆体带严重萎缩,未见明显反射,视网膜厚度中度萎缩。见图2。
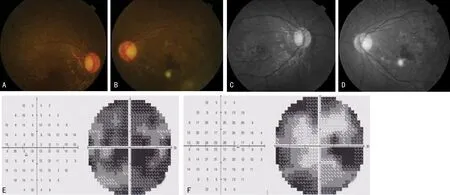
注:A、B为眼底照相,C、D为自发荧光照相,E、F为视野检查;A、C、E示右眼,B、D、F示左眼。

注:A、B为眼底照相,C、D为自发荧光照相,E、F为视野检查,G、H为OCT检查;A、C、E、G示右眼,B、D、F、H示左眼。
3 讨 论
RP1基因于1999年首次被鉴定,它由4个外显子组成,编码2 156个氨基酸[6]。RP1基因编码一种光感受器特异性微管相关蛋白,定位于连接纤毛处,可能参与光感受器内外段之间的蛋白质转运或纤毛结构的维持,在视杆和视锥光感受器外段的结构中起重要作用[7]。
视网膜色素变性包括一组以视杆状细胞为特征的异质性遗传性视网膜营养不良,其特征是视锥细胞变性之前的视杆细胞变性。视网膜色素变性的遗传可以是常染色体显性遗传、常染色体隐性遗传、X连锁隐性遗传或单纯型。迄今为止,已经发现大约80个致病基因与视网膜色素变性相关,其中58个与常染色体隐性遗传形式有关。RP1基因是与视网膜色素变性关联的80多个基因之一,在常染色体显性遗传视网膜色素变性和常染色体隐性遗传视网膜色素变性的病例中已经发现视网膜色素变性相关基因的突变,除了RP1基因,还有bestrophin 1(BEST1)、神经视网膜亮氨酸拉链、核受体亚家族2 E组成员3(NR2E3)、视紫红质(RHO)、视网膜色素上皮基因(RPE65)[8]。RP1基因相关的常染色体隐性遗传视网膜色素变性患者通常比RP1基因相关的常染色体显性遗传视网膜色素变性患者有更严重的视力损害[9]。
本研究纳入的25个家系,全部找到已知或疑似致病突变或临床意义未明的突变,同时,发现RP1基因有9个未报道的新突变。
特别值得注意的是,本研究中患者编号为8号和25号家系的致病突变基因是RP1基因,均指向RP1基因型视网膜色素变性。8号患者RP1基因的c.2886delA和c.4129delG突变为缺失移码突变,属于烈性突变,该突变极可能引起编码蛋白质氨基酸序列发生改变而致病。25号患者RP1基因上有4个致病突变位点,c.4168_4169insT和c.4196delG突变为插入和缺失移码突变,属于烈性突变,而c.4169A>G和c.6353G>A突变为错义突变。经过家系共分离检测确认,笔者发现c.4168_4169insT和c.4169A>G突变位于DNA单链,而c.4196delG和c.6353G>A突变位于另一条链。在临床表现上,笔者也能看到,25号患者的眼底病变程度远比8号患者严重,25号患者黄斑萎缩严重,中心凹处视锥细胞结构消失,自发荧光照相显示色素上皮背景荧光几乎消失,动脉血管萎缩尤其明显。25号患者临床表现出全色盲,考虑与黄斑的发育异常有关。因为与色盲有关的基因如CNGA3与CNGB3等均没有发现异常,而该患者的黄斑发现严重萎缩,视锥细胞的发育缺失,从而导致色觉缺失。而8号患者无色盲,自发荧光照相显示仍有少量色素上皮背景荧光,动脉血管萎缩不明显。综合以上表现笔者认为,RP1基因的多发位点突变导致的视网膜色素变性症状更显著,危害更严重,这在国内其他同类研究中鲜见报道。
已有研究显示,RP1基因的突变可导致不同的临床表型,可以将这些表型根据临床发现、遗传模式、发病年龄和病程在临床上相互区分[10]。基因分析使研究者能够进行有针对性的诊断测试,并确定基因治疗的治疗方法[11]。随着基因治疗等新型治疗手段的出现,识别与RP1基因突变相关的整个临床表型,对于帮助选择合适的患者及评估所提供治疗的效果至关重要。
视网膜色素变性的高发突变基因和突变位点具有异质性,本研究新发现的RP1基因变异位点具有特殊临床表型。但是HUCKFELDT等[12]的研究发现,尽管RP1基因型相同,但临床诊断包括黄斑营养不良(MD)、椎杆营养不良(CRD)和视网膜色素变性;VERBAKEL等[10]的研究也显示,RP1基因的突变可导致不同的临床表型,具体表现为视网膜色素变性、常染色体隐性遗传MD或常染色体隐性遗传CRD,取决于残留的视网膜细胞的数目和生理功能等情况。
RP1基因是与RP关联的80多个基因之一,MIZOBUCHI等[13]的研究表明,RP1基因变异类型/位置和临床表型之间存在基因型-临床表型相关性。RIERA等[14]的研究描述了由RP1基因突变引起的黄斑营养不良表型,并在该基因中建立了新的基因型-表型相关性。本研究所发现这9个RP1基因突变位点,与视网膜色素变性之间及这些相关病例所表现出来的特殊临床表型之间,是否存在必然的联系,还需要通过动物实验、基因敲除、基因修复等进一步的研究来验证。
近年来二代高通量基因测序技术发展迅速,可利用特制探针或基因扩增,对特定的蛋白编码区域DNA或某段特定序列进行目标捕获并富集,进行高通量测序。该技术具有效率高、费用低、时间短等优点,为大量表型复杂的视网膜色素变性家系研究提供了技术支持,是遗传性视网膜色素变性疾病的有效检测手段,有助于研究者发现新发位点突变,并且临床上可以辅助诊断视网膜色素变性类型,提高该类疾病临床诊治能力。此外,对于临床RP1基因治疗,必须全面了解疾病的自然病程,特别是要了解不同的表型,才能较好地评估治疗效果。
