Spin and spin–orbit coupling effects in nickel-based superalloys:A first-principles study on Ni3Al doped with Ta/W/Re
2022-01-23LipingLiu刘立平JinCao曹晋WeiGuo郭伟andChongyuWang王崇愚
Liping Liu(刘立平) Jin Cao(曹晋) Wei Guo(郭伟) and Chongyu Wang(王崇愚)
1Key Laboratory of Advanced Optoelectronic Quantum Architecture and Measurement(MOE),School of Physics,Beijing Institute of Technology,Beijing 100081,China
2Department of Physics,Tsinghua University,Beijing 100084,China
Keywords: nickel-based superalloys, Ni3Al, spin, spin-orbit coupling, mechanical properties, electronic structures
1. Introduction
Recent years have seen the widespread usage of superalloys in turbine engines.[1,2]With excellent creep strength,good oxidation resistance, and good ductility,[3]nickel-based superalloys,i.e.,FCC-Ni(γ)and Ni3Al(γ′),have become popular in the aerospace industry since they have better mechanical properties after doping with certain chemical elements.[4,5]For instance,heavy elements such as Ta,W,and Re are widely used in nickel-based superalloys. The distributions of heavy elements significantly affect the microstructure of the superalloys and are crucial to the performance of nickel-based superalloys.
Many experiments were undertaken to understand the mechanical properties,[6-9]as well as the locations and diffusion of Ta,[10-12]W,[13,14]and Re[15,16]in superalloys. Ta and Re atoms prefer to substitute Al sites inγ′phase,while W atoms show no preference and can appear in bothγand Al sites inγ′phases.[14]Doping methods have been applied in superalloys to increase their ductility.[17]However, it is difficult to prepare a high-quality,heavy-element-doped superalloy,and it can be challenging to locate the optimal position of the heavy metal atoms in such a superalloy given its complicated microstructures. Theoretical simulations are helpful in decreasing the cost of superalloy studies. There are some theoretical calculations explaining the experiments,[10]showing where heavy elements prefer to reside,[18,19]and the reason whyγandγ′phases with chemical defects have good mechanical properties.[20-23]The mechanism of solid solution strength in superalloys remains elusive since accurate simulations with numerous species of heavily metallic elements(typically with ten species) are difficult. Moreover, considering the spin degree of freedom in simulations of systems with hundreds of metallic atoms is computationally demanding, and the spinorbit coupling (SOC) effect is rather challenging for systems with hundreds of atoms. It is difficult to describe the magnetism of Ni3Al due to spin-flucuations.[24]Although Ni3Al is a weak ferromagnet and has a low Curie temperature(41 K),doping 5d elements may introduce local moments in the crystal and exibite different magnetic properties, especially when the alloys are produced and cooled to room temperature. For heavy atoms, the SOC effects can yield a large difference in predictions of likely electronic structures. Up to now, the SOC effects are rarely considered in published work related to superalloys. Importantly,systemic investigation of the roles played by spin and SOC in determining the lattice constant,space group symmetry, dopant formation energy, as well as magnetic and mechanical properties, is warranted, especially in Ni-based superalloys.
In the present research,by performing first-principles calculations, we have systematically calculated the lattice constant,formation energy,mechanical properties,and electronic structures considering spin and SOC effects in FCC-Ni(γ)and Ni3Al(γ′) and their Ta, W, and Re-doped alloys, then compared them with non-spin polarized results. We found that including spin for bothγandγ′phases is necessary and sufficient in most cases, except that dopant formation energy is sensitive to different spin effects;even spin-polarized calculations give 1% to 9% variance compared to SOC at a doping density of 1/32.
2. Method and model
First-principles calculations were performed by using Viennaab-initiosimulation package (VASP) with projectoraugmented-wave (PAW) potentials.[25-27]The exchangecorrelation interactions are treated within the generalized gradient approximation (GGA) in the scheme of the Perdew-Burke-Ernzerhof(PBE)functional.[28]The energy cut-off for plane-wave basis is set to 400 eV.The Brillouin zone is sampled by a uniformed grid (9×9×9) for the unit cell of Ni3Al, and the grid density is kept fixed in supercell calculations. First-order Methfessel-Paxton smearing is used in selfconsistent field calculations.[29]
The SOC effect is derived from the relativistic Dirac equation under the low-velocity approximation, where SOC term can be written as

wherea0is the Bohr radius,Zrepresents the number of nuclear charges,nis the main quantum number,lis the angular quantum number, andjis the total angular quantum number(J=L+S). So, in the spherical symmetry approximation,the SOC strength is proportional toZ4. As a result,heavy elements with largeZshow strong SOC effects. The inversion and the time reversal symmetries of the Ni3Al system with doping atoms are broken. It will induce a Zeeman splitting,and,the energy scale of this splitting is governed by the SOC strength. Given the heavy doping atom,it suggests that a pure non-spin calculation is inappropriate for such systems.
The conjugate gradient(CG)algorithm is used for structural relaxation. Volumes and bulk modulus can be calculated using Rose-Vinet[32]and Birch-Murnaghan[33,34]equations of states. In this work, the Birch-Murnaghan equations of states are used to optimize the volumes and calculate the bulk modulus of the structures:[34]these were processed by the Atomic Simulation Environment (ASE) package.[35]Forces on each atom are relaxed with a convergence criterion of 0.01 eV/˚A.

Fig.1. (a)Ni(γ)unit cell(b)Ni3Al(γ′)unit cell;(c)2×2×2 supercell of Ni3Al, and one of the Ni atoms is substituted by a doping atom X,where X =Ta,W,or Re;(d)2×2×2 supercell of Ni3Al,and one Al is substituted by a doping atom X.
The strain tensor can be written in the following form:
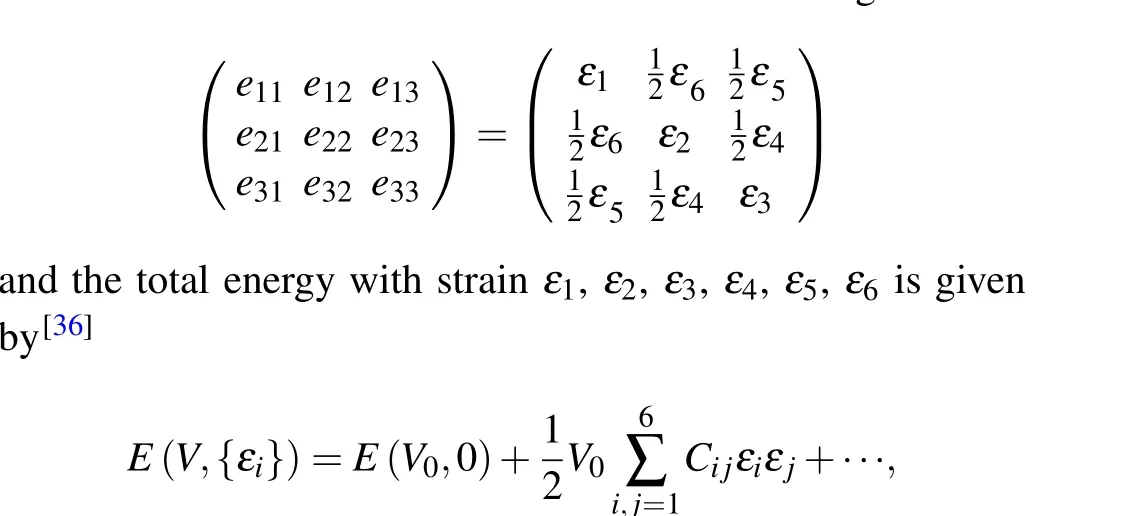
whereCijdenotes the elastic constants, as calculated by use of the second-order derivative of total energy,[37,38]E(V,{εi})andE(V0,0) are the total energy with and without strain, respectively, andV0represents the volume at equilibrium state.Five different strains from-1.0% to 1.0% are applied to the structures to fit theE-εcurve. The formation energy calculation is based on the Wagner-Schottky model,[39]as stated below(a more negative value means more energy-favorable),and all total energies are calculated using the same parameters andk-mesh density:

Python Materials Genomics(pymatgen)[40]and vaspkit[41]are used to post-process electronic structure calculations. The unit cells of FCC-Ni(γ)and Ni3Al(γ′)are shown in Figs.1(a)and 1(b). Considering the fact that the doped heavy metal in Ni3Al is a few percent in mole fraction in experiment, we have used a 2×2×2 Ni3Al supercell to simulate a heavy atom substitutional defect by replacing Ni/Al atom with anX(X=Ta/W/Re)atom as illustrated in Figs.1(c)and 1(d).
3. Results
For models containing heavy atoms of Ta/W/Re,we treat SOC results as “correct”, and compare results with non-spin and spin-polarization with SOC systematicallly in lattice constant,bulk modulus,elastic constant,dopant formation energy and electronic structures. As shown in supplementary material Table S1, our calculated lattice constant, bulk modulus,and elastic constant of Ni3Al agree with previous calculations and experiments.
3.1. Lattice constants
We have calculated the lattice constants of theγ,γ′phases with and without substitutional defects. Considering SOC as a“standard”calculation,non-spin and spin-polarized calculations exhibit different behaviors. SOC calculations give lattice constants of 3.518 ˚A and 3.567 ˚A forγandγ′phases,respectively. The non-spin calculation gives a lattice vector that is 0.007 ˚A and 0.002 ˚A smaller forγandγ′,respectively.As shown in Fig.2(a),spin polarized calculations give almost the same lattice parameters as SOC since the SOC effects are weak for light atoms.The spin polarized calculations are sufficient for Ni(γ)and Ni3Al(γ′)lattice relaxations. In Ni3Al(γ′),for high density of Re-dopedγ′(1×1×1 unit cell),non-spin introduces a difference of less than 0.1%;for the case of a low doping density(2×2×2 supercell),the difference is less than 0.03%,as shown in Fig.2(b). Thus,spin and SOC effects are negligible when calculating lattice constants with a low doping density in Ni3Al(γ′).
Heavy-atom dopants enlarge the lattice of theγ′2×2×2 supercell by 0.01 ˚A to 0.06 ˚A as shown in supplementary material Table S2. Among Ta,W,and Re in theγ′system,due to the fact that the atomic radius of Ta is greater than that of W and Re, Ta has the biggest lattice at about 7.192 ˚A, which is 0.81%larger than theγ′supercell itself;W and Re enlarge the lattice by about 0.35%. The substitution of TaNiincreases the lattice constant by about 0.8%compared to the substitution of TaAl.

Fig.2. Difference in lattice constants from non-spin,spin,and SOC calculations. (a)FCC-Ni and Ni3Al lattice,(b)percentage difference of non-spin and spin lattice constant with respect to SOC calculations.

Fig. 3. Difference in bulk modulus from non-spin, spin and SOC calculations. (a)FCC-Ni and Ni3Al bulk modulus,first four groups are Ni and Ni with defects in the γ phase,and others are Ni3Al and Ni3Al with defects in the γ′ phase. (b)Bulk modulus of doped structures in the γ′phase with SOC,the Re-replacing-Ni case gives a larger bulk modulus(by about 6 GPa)than Re/W-replacing-Ni.
3.2. Bulk modulus
We have also calculated the bulk modulus of all the systems in the 2×2×2 supercell. As shown in Fig.3,non-spin and spin-polarized calculations often give larger values than SOC,except for ReNiinγandγ′. Besides, non-spin calculations may introduce a variance of up to 5 GPa(3%)compared to SOC,but the value for spin-polarized cases is mainly around 1%. Interestingly, the Re atom inγ′results in an increase of bulk modulus of 6 to 7 GPa,showing that Re is more efficient than Ta and W in terms of increasing the bulk modulus.
3.3. Formation energy
Formation energy is an important parameter to determine whether and where a dopant locates in superalloys. We have calculated the formation energy of Ta, W, and Re replacing Ni or Al in theγ′phase of a 2×2×2 supercell at a doping density of 1/32. In Ni3Al,it is found experimentally that substitutional Ta and Re atoms prefer to locate at Al sites,[10,13]which is consistent with our calculations(supplementary material Table S3). Our experimental results in the National Key Research and Development Program also gave the same conclusion. As shown in Fig.4,compared to the SOC calculated formation energy,non-spin calculations give a large difference(about 10 to 30%),and spin-polarized calculations improve the results,with differences of less than 10%with respect to SOC.Such difference implies that at doping density as low as 1/32,the SOC effects are still remarkable when calculating defect formation energy. For larger systems with several hundreds of atoms, we suggest that spin-polarized calculations should be performed.

Fig.4.Formation energies from non-spin,spin-polarized,and SOC calculations. The percentage is calculated with respect to SOC results of the supercells(2×2×2)of the γ′ phase.
3.4. Elastic constant
To estimate the potential spin and SOC effects on the mechanical properties of superalloys,we have also calculated the elastic constant in the 2×2×2 supercell of theγ′phase(C11shown in Fig.5 andC12in supplementary material Table S5)considering spin and SOC effects and compared them with non-spin results. The trend is that, spin-polarized results are closer to SOC;however,as we increase the SOC strength manually to 200% and 300%, they approach the non-spin results again. The reason for this can be seen from the electronic calculations shown below,and we conclude that for large systems with several hundreds of atoms,the spin-polarized calculations should suffice.

Fig. 5. Elastic constant C11 under non-spin, spin-polarized, and SOC calculations. The data are shown in supplementary material Table S4.
3.5. Electronic structure calculations
To investigate the mechanism of the overestimations of the bulk modulus, taking the Re doped 2×2×2 Ni3Al as an example,we performed differential charge density(DCD)calculations as shown in Fig. 6. The DCD represents the redistribution of charge density upon interactions between atoms,which is calculated as the following equation:

Here,Ψis the wavefunction in DFT calculations,andρis the density of electrons. Positive values (density of electrons increased)are colored in red,and negative values are in blue.
For the non-spin result, a significant charge transfer can be observed from the Ni atom to the doped Re atom, resulting in a strong dopant formation energy. By contrast, in the spin-polarized and the SOC results,the charge density,which comes from both Ni and Re atoms, aggregates between the two atoms,resulting in a relatively weak metallic bond. Thus,a relatively small bulk modulus should be observed,as shown in Fig. 3. The weakening of the metallic bond in the presence of SOC can be understood as follows. The d orbitals of the heavy doping atoms can give rise to an effective magnetic field that couples with the spin magnetic moment, i.e.,-µ·B. Here,µis the spin magnetic moment, andBis the magnetic field of the orbital moment. As a result, the spin polarization tends to align to the effective magnetic field. To form a chemical bond, it may cost additional energy to break the alignment comparing with the case without SOC.Despite of our simplified model, the argument of the softening of the chemical bond as well as the bulk modulus has its general validity in Re doped Ni3Al system. For the typical concentration of the heavy doping atom, our results suggest that the SOC softens the chemical bond of its vicinity, giving a change in the bulk modulus. Of course,how strong SOC will affect the bulk modulus is dependent on the relative strength of the SOC compared to the crystalline electrostatic field and the dopant density.
We calculate the band structures of 2×2×2 supercell(Ni24Al7X). Since the bands are folded in the Brillouin zone of the supercell, we perform a band unfolding calculation by vaspkit.[41]The unfolded non-spin, spin-polarized and SOC band structures of Ni24Al7Re, Ni24Al7Ta, and Ni24Al7W are shown in supplementary material Figs. S1-S3. In Fig. 7, we take Ni24Al7W as an example. We can see that the bands nearΓpoint and Fermi level are spilt in spin-polarized calculations,and the SOC effect tends to close the band gap of spin up and down,which causes the differences of the total energies in non-spin,spin-polarized,SOC calculations,and further varies the relevant properties.
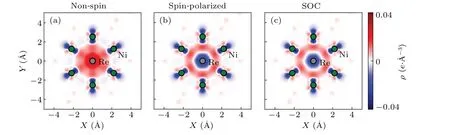
Fig. 6. Differential charge density analysis of Ni24Al7Re, from (a) non-spin, (b) spin-polarized and (c) SOC calculations, respectively. The electrons are transformed from the blue region to the red region. In contrast with the non-spin calculations,the spin-polarized and SOC ones have fewer electrons transforming from the Ni atoms to the Re atom,indicating the intensity of the metallic bonds is overestimated by the pure non-spin calculations. The reasons are discussed in the main text.

Fig.7. (a)Unfolded band structure of Ni24A7W,non-spin calculation;(b)local zoom near Γ point and Fermi level of unfolded band structure.The scattering size indicates the weight of unfolded effective band structure in folded band structure.

Fig.8. Density of states by non-spin,spin,and different SOC strength calculations. The Fermi level has been set to 0. Here SOC300 means the SOC effects are artificially enhanced to 300%.
To highlight the SOC effect,we have also performed electronic structure calculations in the unit cell,using Ni3W as an example for all the density of states (DOS), band structure,and magnetic moment calculations. Four types of calculations are performed, which are non-spin, spin-polarized, SOC and 300% SOC. DOS of Ni3Re and Ni3Ta are shown in the supplementary material. For the DOS shown in Fig.8,non-spin,spin-polarized and SOC calculations agree with each other except for some energy ranges near the Fermi level. Besides,spin-polarized calculations cause a split near-2.5 eV due to the spin split on d band of Ni, according to the projected local DOS of d band shown in supplementary material Fig.S4.The t2gorbitals cause the greatest effect at-2.5 eV; both t2gand egorbitals split near the Fermi level due to the spin polarized effect,as show in supplementary material Fig.S7. Moreover, SOC effects on the d state of heavy metal further split the spin-up and spin-down states. This is more obvious for the calculations with 300%-SOC (see Fig. 8(b)). To understand the details,the band structures are analyzed.
In Fig. 9, the spin and SOC effects are shown to split the bands of the non-spin calculations, which is most obvious from pathRtoΓ. For bands dominated by the d state of heavy metal W, both spin-up and spin-down bands further split slightly due to the SOC effect, resulting in a decreased spin-up and spin-down separation in energy scale, and the magnetic moment of Ni3W is decreased from 1.8µB(spinpolarized result)to 1.3µB(SOC result),and the stronger SOC(300% SOC) recreates degenerate states and causes the disappearance of the magnetic moment of the system. So, SOC effect tends to reduce the total magnetic moment of the alloys,as shown in Table 1. We can also find that the bands near the Fermi level of pathX-M-Rin non-spin calculation move upward and converge at theR-point in the 300% SOC strength calculation, contributing to the DOS peak-shift at the Fermi level shown in Fig.8.
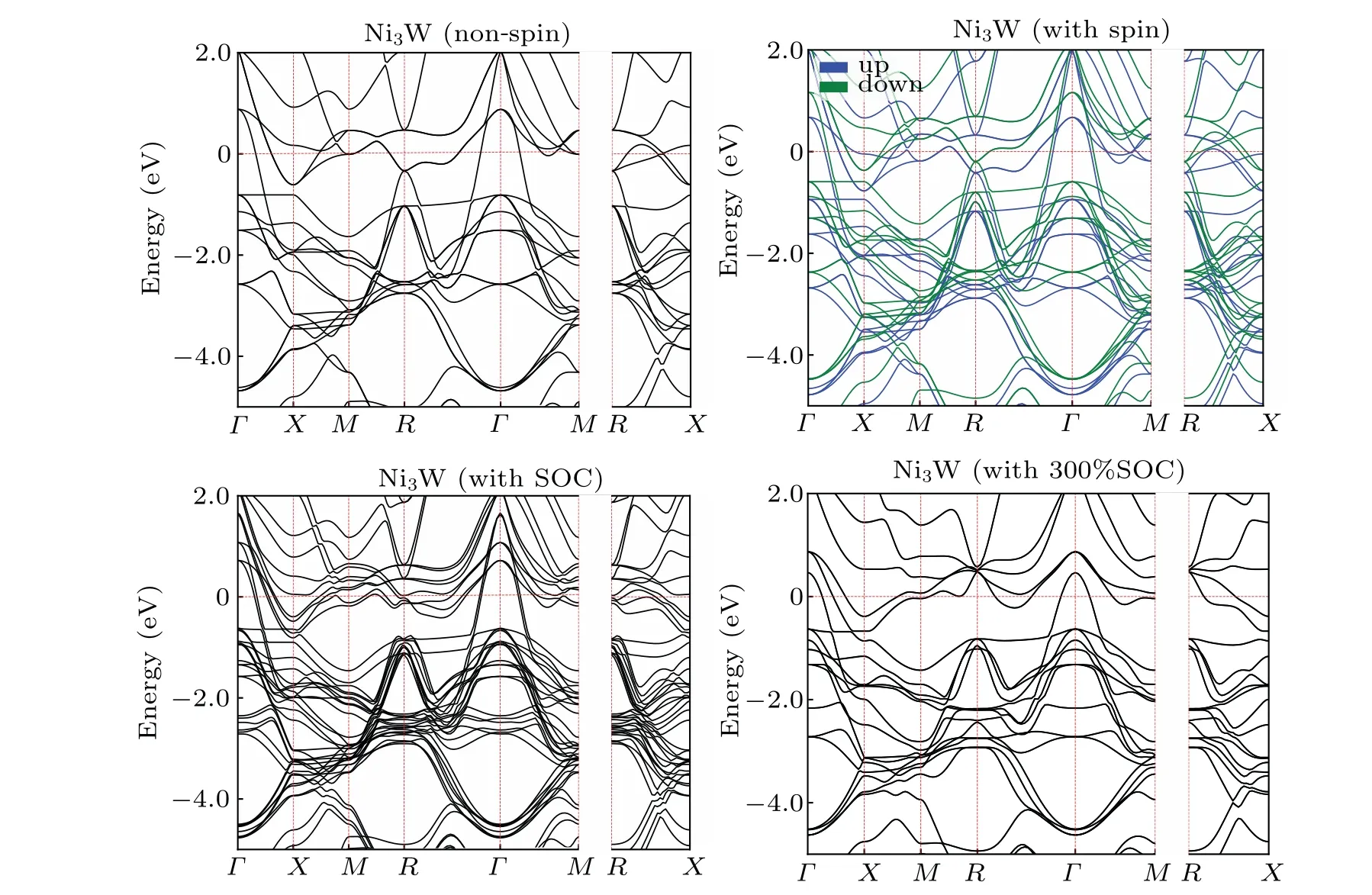
Fig.9. Band structure of Ni3W under different spin effects. The Fermi level has been set to zero. All of the high-symmetry lines are included in band-structure calculations,which follows the convention of AFLOWLIB.[42]
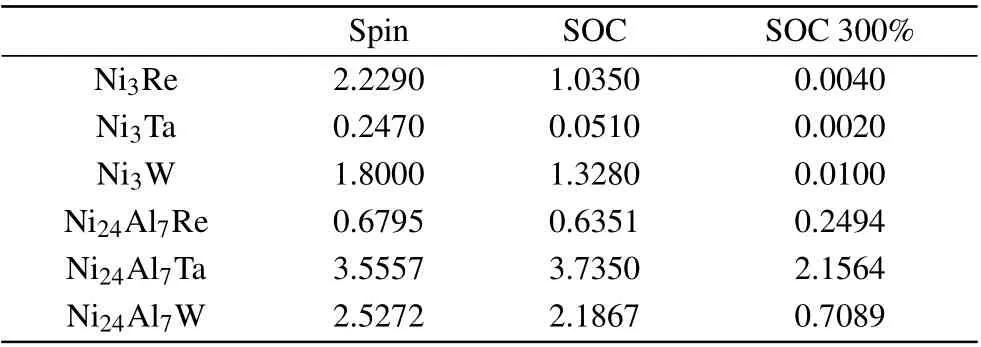
Table 1. Magnetic moments for Ni3Re, Ni3W, Ni3Ta, Ni24Al7Re,Ni24Al7Ta,and Ni24Al7W in units ofµB.
4. Conclusion
The lattice constant,formation energy,mechanical properties, and electronic properties ofγ,γ′, and their Ta, W,and Re-doped alloys are calculated. Results under non-spin,spinpolarized, and SOC effects are compared. The lattice constant is not sensitive to the SOC effects and spin polarized calculations suffice for theγphase. In theγ′phase, spin and SOC effects are negligible when calculating the lattice constant. For the bulk modulus, the difference can be between 1% and 3% without the SOC effect, and spin-polarized calculations can reduce the difference to 1%,so,we recommend including spin effect for bothγandγ′phases. Dopant formation energy is sensitive to different spin effects,even spinpolarized calculations give difference of between 1%and 9%at a doping density of 1/32, which make us aware of the difficulty of finding where heavy atoms are located in theγandγ′phases. The SOC effects on the elastic constant are small,at less than 5%at 1/32 dopant density,for large systems,spinpolarized calculations should suffice. Magnetic moments are also affected by different strengths of SOC calculations: a high SOC strength decreases the total magnetic moments to a greater extent. Electronic structures calculations indicate that spin polarization causes a split in the metal d states,and SOC introduces a further split in the spin-up and spin-down states of the d states of heavy metals and reduces the magnetic moment of the system. To sum up, spin-polarized calculations suffice for most of the properties mentioned in this study except for dopant formation energies, which can be as large as a few percent at a 1/32 dopant density. This makes it difficult to predict the heavy metal distribution in superalloys,since including SOC effect in structural relaxation is quite expensive.
Acknowledgement
Project supported by the National Key Research and Development Program of China (Grant Nos. 2017YFB0701603 and 2017YFB0701502).
杂志排行

Chinese Physics B的其它文章
- Superconductivity in octagraphene
- Soliton molecules and asymmetric solitons of the extended Lax equation via velocity resonance
- Theoretical study of(e,2e)triple differential cross sections of pyrimidine and tetrahydrofurfuryl alcohol molecules using multi-center distorted-wave method
- Protection of entanglement between two V-atoms in a multi-cavity coupling system
- Semi-quantum private comparison protocol of size relation with d-dimensional GHZ states
- Probing the magnetization switching with in-plane magnetic anisotropy through field-modified magnetoresistance measurement