火焰喷雾合成法制备La0.8Sr0.2Mn1-xCuxO3催化氧化CO性能研究
2021-10-31周昊伍其威程方正
周昊,伍其威,程方正
(浙江大学能源清洁利用国家重点实验室,浙江杭州310027)
引 言
CO是一种无色、无味、无刺激性的有毒大气污染物,主要在燃烧过程中产生。煤炭、石油和天然气等传统化石燃料燃烧时,其中烃类和含碳物质的不完全燃烧是CO的主要来源。汽车发动机尾气、废弃物焚烧及电站锅炉排放废气中同样含有大量CO[1]。以钢铁工业为例,规定了烧结烟气中CO排放浓度不得超过4000~5000 mg/m3[2]。CO未经脱除排放不但会对环境造成危害,同时CO极易与血红蛋白结合,浓度过高时会损害人体神经系统,引发安全事故[3-4]。因此,对排放废气中CO气体进行有效脱除至关重要。现有诸多治理方法中,通过高效催化剂将CO催化氧化成CO2被视为最有效的脱除技术,受到广泛关注。在CO催化氧化研究中,贵金属催化剂(如Ru、Pt、Au)具有良好的催化活性,但存在容易烧结,价格昂贵,且活性随反应时间而降低等诸多缺点[5-6]。同时,工业排放废气中通常含有大量水蒸气和CO2等杂质气体,会影响催化剂催化性能,导致催化剂失活[7]。相较于传统的贵金属催化剂,钙钛矿型氧化物对CO催化氧化性能优异,具有丰富的组成灵活性和较高的结构可调性,在环境能源领域被视为一种极具潜力的高效催化剂。
钙钛矿是一类具有ABO3特定晶格结构的氧化物,其A位元素主要为碱金属或碱土金属,如La、Ca等,而B位元素多为过渡金属元素,如Ni、Co、Mn等。其中,A位金属元素主要起稳定钙钛矿晶体结构作用,而B位元素的类型及其氧化价态循环难易直接影响催化剂活性,对CO催化氧化性能起到至关重要的作用[8-10]。此外,通过A、B位阳离子的部分取代来改变元素化合价和氧的非化学计量关系,可实现其氧化还原性能提升。根据之前的研究报道,LaMnO3钙钛矿常用于CO催化氧化,通过A、B位阳离子部分取代后能获得较好的CO催化性能[11]。Frozandeh-Mehr等[12]制 备 了A位 部 分 取 代 的La1-xMxMnO3(M=Sr或Bi,0≤x<0.4)钙钛矿,结果表明,Sr和Bi掺杂对LaMnO3钙钛矿相形成影响较小,且对降低CO氧化温度具有积极作用。Teng等[13]通过水热法和柠檬酸法分别合成了La0.5Sr0.5MnO3钙钛矿,并研究对比了合成方法对CO的催化氧化性能影响,发现在220℃以下即可实现CO完全催化氧化。Kucharczyk[14]分别合成了A位和B位部分取代的La1-xPdxMnO3和LaMn1-xPdxO3钙钛矿,催化剂对CO的活性随Pd掺杂量的增加而提升。钙钛矿B位取代可提供变价金属离子,其价态循环难易程度决定了钙钛矿氧化物的催化活性[15]。Wu等[16]通过溶胶-凝胶法制备了B位部分取代的LaM0.25Co0.75O3(M=Cu,Mn,Fe)钙钛矿,探究其对CO催化还原NO的催化性能,结果表明Cu的部分取代促进了钙钛矿还原能力和吸附能力,其中LaCu0.25Co0.75O3催化剂具有最佳的活性和长期催化稳定性。Tarjomannejad等[17]研究了溶胶-凝胶法合成的钙钛矿型氧化物LaMn1-xBxO3(B=Cu,Fe;x=0,0.1,0.3,0.5)对CO的催化氧化作用,研究发现,Cu取代的钙钛矿具有更高的CO氧化活性,这与Cu和Mn之间的协同作用有关,在Cu掺杂量为0.3时具有最佳的催化活性。由于钙钛矿结构的灵活可调性,可对A、B位阳离子同时部分取代,形成AA′BB′O3型氧化物。Huang等[18]采用熔盐法制备了多孔La0.8Sr0.2Mn1-xCuxO3和La0.8Sr0.2MnO3纳米粒子,对比了钙钛矿结构在B位掺杂Cu2+条件下CO转化率,结果显示Cu2+能有效提高催化剂的还原能力,LSMC-0.2具有最佳的催化活性。因此,本文选择LaMnO3型钙钛矿催化剂,并通过Sr2+、Cu2+分别部分取代A位和B位。
现有报道的用于CO催化氧化的高效钙钛矿型催化剂大多采用溶胶-凝胶法[16-17]或水热法[19]等传统化学液相合成法,在实际制备过程中,常存在制备周期时间长、操作较复杂、合成效率低等问题。近年来,火焰喷雾合成法作为一种典型的高温气相合成纳米粒子技术,相比传统的化学液相合成法合成纳米颗粒,具有操作简单、反应时间短、合成效率高等特点[20-21],并逐渐应用于工业生产实践。在火焰喷雾合成过程中,一般把所需的盐类化合物或金属配合物作为前体雾化后通入火焰,由高温火焰点燃,并在较短时间内经历反应、成核和烧结等复杂的“液滴-颗粒”转换过程,最终形成目标纳米颗粒[22]。Abe等[23]制备了具有高结晶度和高比表面积的钙钛矿型纳米粒子La2TiO7,火焰喷雾合成法制备的催化剂优化了光催化性能,适合制备具有特定形貌和精确化学组成的纳米钙钛矿催化剂。Kim等[24]利 用 火 焰 喷 雾 法 合 成 了La0.2Sr0.8Co1-xFexO3-δ和Ba0.5Sr0.5Co1-xFexO3-δ纳米钙钛矿,催化剂析氧反应的活性和稳定性大大增强。因此,火焰喷雾合成法具有合成高效钙钛矿催化剂的应用潜力。
本文通过火焰喷雾合成法制备了La0.8Sr0.2Mn1-xCuxO3(x=0,0.1,0.2,0.3,0.4)钙钛矿催化剂用于CO催化氧化实验。选择Sr2+和Cu2+作为掺杂元素,同时部分取代LaMnO3的A位和B位,对火焰喷雾法合成的催化剂进行XRD、SEM、EDS、BET、H2-TPR和O2-TPD等测试手段进行表征。研究比较了La0.8Sr0.2Mn1-xCuxO3催化剂对CO催化氧化的活性。模拟了实际工况中含水蒸气和CO2混合气的测试条件,探究催化剂在水蒸气和CO2环境下的CO氧化性能。最后还测试了不同掺杂比例钙钛矿的连续催化稳定性和活性。
1 实验部分
1.1 催化剂制备
火焰喷雾法合成制备La0.8Sr0.2Mn1-xCuxO3(x=0,0.1,0.2,0.3,0.4)钙钛矿催化剂系统如图1所示。按一定摩尔比称取La(NO3)3·6H2O(≥99.9%)、Mn(NO3)3·4H2O(≥98%)、Cu(NO3)2·H2O(≥99.7%)和Sr(NO3)2(≥99.5%),溶解于无水乙醇(≥99.7%)中,配制成0.2 mol/L金属离子浓度的前体溶液,同时分别控制摩尔比例n(Sr)∶n(La)=1∶4,n(Cu)∶n(Mn)=1∶9、1∶4、3∶7和2∶3。微流注射泵(Harvard Pump 11 Elite)控制前体溶液以速率为1.2 ml/min进入燃烧器喷嘴,同时通入0.5 L/min的空气,形成稳定的前体喷雾。雾化后的液相前体由Mckenna燃烧器的平面支撑火焰点燃(2 L/min CH4+20 L/min空气)。前体在高温火焰中反应,进行成核、生长和烧结等过程,最终形成的钙钛矿颗粒产物伴随高温气体由玻璃纤维滤膜(Whatman GF/D)配合真空泵收集。
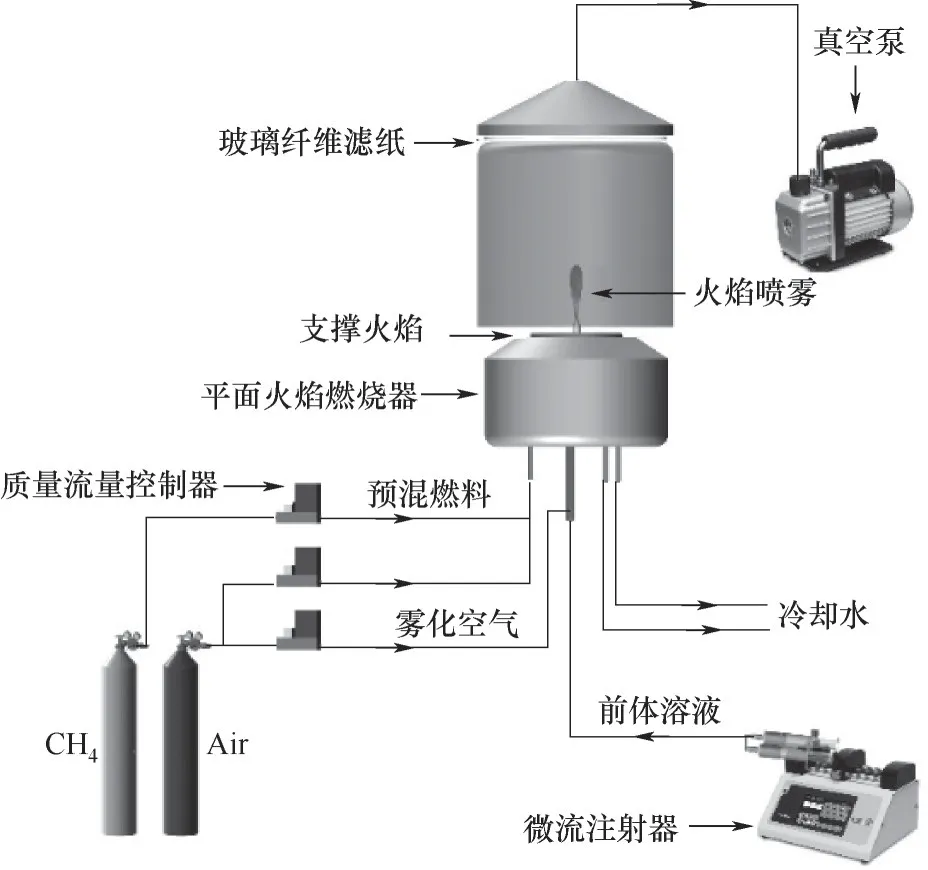
图1 火焰喷雾合成法实验系统Fig.1 Scheme of the flame spray synthesis system
1.2 催化剂表征
通过X射线衍射(X-pert Powder,PANalytical B.V.)测定钙钛矿产物的相成分。利用单色Cu Kα1辐射(λ=0.154 nm)X射线在2θ=20°~80°范围之间按0.02(°)/s连续扫描。钙钛矿微观形貌通过日立SU-70场发射扫描电镜(Hitachi SU-70 Analytical Field Emission Scanning Electron Microscopy,FE-SEM)观察,配合能量色散X射线能谱(energy dispersive Xray spectroscopy,EDS)测定了样品的体成分。表面元素价态分析(XPS)采用Thermo Kalpha光电能谱仪对材料表面元素分布及价态进行表征测试,并采用284.8 eV的外源C 1s对XPS谱图进行校正。将0.5 g样品在100℃下预处理10 h后在623 K下脱气2 h,最后在77 K下通过N2吸附BET法确定钙钛矿的比表面积(specific surface area,SSA)。H2程序升温 还 原 测 试(temperature programmed reduction,TPR)在全自动化学吸附仪(BelCata II,Japan)上进行。首先将50 mg样品在150℃下预处理1 h,温度降至室温后再以30 ml/min通入10%(体积)H2/Ar,并以10℃/min升温速率由室温升至900℃,同时通过热导检测器(thermal conductivity detector,TCD)记录信号得到TPR曲线,并检测H2消耗量。O2-TPD分析在全自动化学吸附仪(BelCata II,Japan)上进行,首先将100 mg样品在400℃以流量为30 ml/min的O2预处理1h,降至室温吹扫30 min,切换至He,待基线稳定后以10℃/min升温速率由室温升至900℃,同时通过TCD记录信号得到样品TPD曲线,并检测O2的脱附量。
1.3 CO催化氧化实验
催化剂性能测试实验装置如图2所示,两种测试工况见表1。催化氧化CO的活性测试在石英管固定床反应器中进行(内径25 mm,长度500 mm),采用精度为±0.5%的Alicat质量流量计分别控制反应原料气体积分数组成为1%CO+10%O2+89%Ar,控制总气流量为1.5 L/min。反应器在管式炉内以6℃/min从室温程序升温加热至300℃,并通过气体分析仪(Testo 350)测定记录CO浓度随温度的变化。
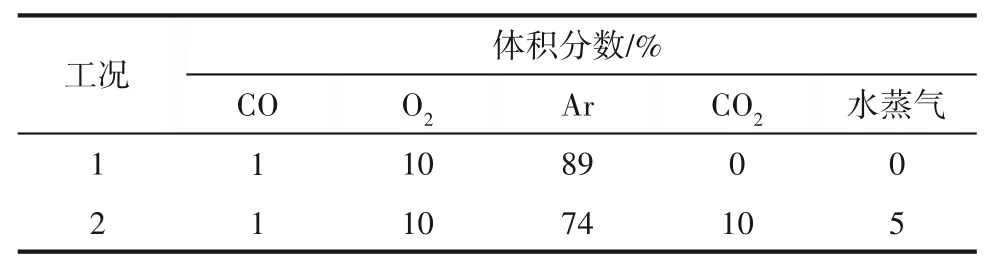
表1 La0.8Sr0.2Mn1-xCuxO3(x=0,0.1,0.2,0.3,0.4)催化剂实验工况Table 1 Experimental conditions for catalyst performance test
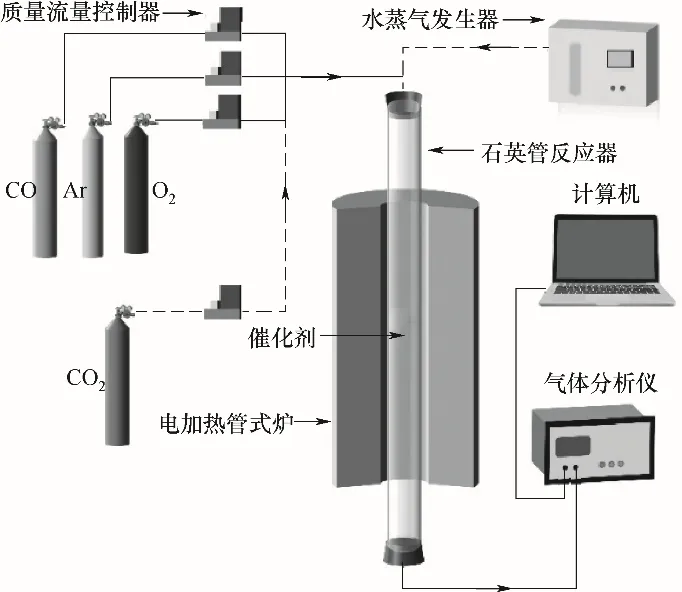
图2 固定床钙钛矿催化氧化CO实验系统Fig.2 Fixed bed system for catalytic oxidation of CO with perovskite catalysts
混合气体测试中,反应原料气体积分数组成为1%CO、10%O2、10%CO2、5%水蒸气和74%Ar。待稳定后,从室温程序升温至300℃,并配合气体分析仪测定记录CO含量变化。同时为探究火焰喷雾合成催化剂对CO催化氧化的长效稳定性和活性变化,对每种催化剂的两种工况分别进行了5次连续催化氧化实验。测试条件保持一致,得到CO转化率为50%和90%时的对应温度,CO转化率通过式(1)计算:
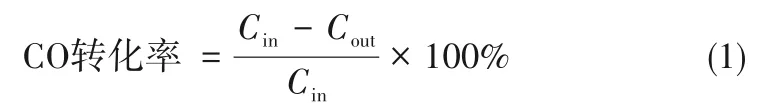
式中,Cin为通入混合气中CO浓度;Cout为反应后出口CO的浓度。
2 结果和讨论
2.1 催化剂的表征
La0.8Sr0.2Mn1-xCuxO3(x=0,0.1,0.2,0.3,0.4)钙钛矿催化剂的XRD结果如图3所示。由图可以看出Sr取代量为0.2时,在不同Cu浓度取代条件下,催化剂均呈现良好的LaMnO3钙钛矿相(PDF# 72-0440),并且具有较好的结晶度,说明火焰喷雾合成法制备了较理想的钙钛矿型氧化物。从XRD衍射图结果可看出,La0.8Sr0.2MnO3中出现8个明显的衍射峰,分别对应2θ为22.8°、32.4°、40.1°、46.6°、52.5°、58.0°、68.1°和77.6°。当x≤0.2时,催化剂的XRD衍射图像基本与La0.8Sr0.2MnO3保持一致,说明低浓度的Cu2+掺杂能有效进入钙钛矿晶胞结构中,同时保持结构稳定。而价态较低的Cu2+取代进入晶格后,电荷平衡要求会导致Mn3+向更高价态转变,同时促进催化剂表面活性氧空位的产生[25]。
随着Cu2+掺杂浓度进一步提高,当x≥0.3时,XRD衍射峰显示催化剂中开始出现CuO和La2CuO4相,这表明Cu2+取代Mn3+导致钙钛矿结构稳定性下降,不利于钙钛矿纯相的保持,部分Cu2+开始脱离钙钛矿晶格以其他相形式存在,这与之前研究报道一致[26]。当取代量x=0.4时,La0.8Sr0.2Mn0.6Cu0.4O3中各衍射峰强度已降至最低,说明钙钛矿结构逐渐破坏,样品结晶度下降,过量的Cu掺杂也可能不利于催化剂催化性能的提高。从XRD衍射局部放大图3(b)可以看出,在2θ=32.4°处的衍射峰随Cu2+掺杂量的增加逐渐向更低的2θ角度移动,这是由于Mn3+(n=0.0645nm)在被较大的Cu2+(n=0.0730nm)部分取代后,导致钙钛矿晶格的延伸扩大,体积膨胀,同时会增加氧空位数量[18],Cu元素取代对催化剂的晶型结构特征参数有一定影响。
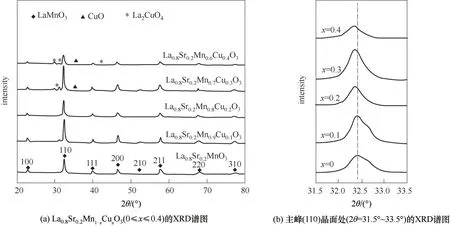
图3 钙钛矿催化剂的XRD谱图Fig.3 XRD patterns of perovskite catalysts
图4为La0.8Sr0.2Mn1-xCuxO3(x=0,0.1,0.2,0.3,0.4)的FE-SEM图像。从图中可以看出,所有催化剂微观结构很相似,没有发生显著的变化,均表现为球状颗粒团聚成的疏松多孔结构。La0.8Sr0.2Mn1-xCuxO3合成所需的前体溶液主要通过硝酸盐溶解于无水乙醇得到,根据此前研究,以硝酸盐为前体的反应物,硝酸根在高温条件下会热解并产生大量气体,进而对制备的催化剂形貌产生重要影响[20,27]。因此,推测这样的疏松多孔结构可能是由于制备过程中前体溶液中的硝酸根在高温火焰中反应分解,产生的溢出气体造成的,有利于增加催化剂的比表面积,提高催化剂活性位点数量,对催化活性提高有积极促进作用[28]。

图4 La0.8Sr0.2Mn1-xCuxO3的FE-SEM图Fig.4 FE-SEM images of La0.8Sr0.2Mn1-xCuxO3
随着Cu2+掺杂比例的增加,催化剂表面开始出现一些细微的颗粒。对于La0.8Sr0.2Mn0.6Cu0.4O3催化剂,其表面出现更多小颗粒且团聚现象越来越明显。结合XRD相成分分析,推测这些小颗粒成分可能为CuO和La2CuO4等。五种钙钛矿型催化剂La0.8Sr0.2Mn1-xCuxO3(x=0,0.1,0.2,0.3,0.4)通过EDS确定的催化剂体成分金属原子百分比见表2。以La0.8Sr0.2Mn0.9Cu0.1O3为例,其EDS结果如图5所示,可以看到催化剂内各元素分布均匀,说明火焰喷雾法制备的催化剂体成分符合预期。
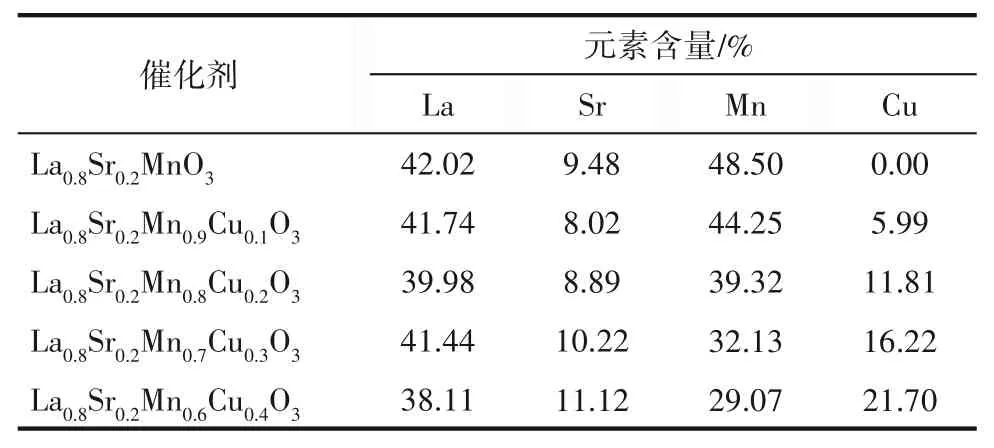
表2 La0.8Sr0.2Mn1-xCuxO3(x=0,0.1,0.2,0.3,0.4)EDS结果Table 2 EDS results of La0.8Sr0.2Mn1-xCuxO3(x=0,0.1,0.2,0.3,0.4)

图5 La0.8Sr0.2Mn0.9Cu0.1O3和La0.8Sr0.2Mn0.6Cu0.4O3的EDS能谱图Fig.5 EDS mapping of La0.8Sr0.2Mn0.9Cu0.1O3 and La0.8Sr0.2Mn0.6Cu0.4O3
La0.8Sr0.2Mn1-xCuxO3(x=0,0.1,0.2,0.3,0.4)钙钛矿催化剂的比表面积测试结果见表3。在La0.8Sr0.2Mn1-xCuxO3钙钛矿中,样品平均比表面积为9.85~17.68 m2/g,最大标准差约为5%。随Cu2+的部分取代,比表面积逐渐增大,并在x=0.4时达到最大值。这说明钙钛矿B位Mn3+被Cu2+部分取代后,比表面积会逐渐增大,这与之前研究报道一致[29]。结合XRD和SEM结果分析,当Cu掺杂量增加时,推测比表面积的增大可能是由于Cu的引入促进了颗粒粒径减小,同时出现CuO和La2CuO4相的分离,形成更多微小颗粒,并在表面富集,使其具有更高的比表面积。

表3 BET法测定La0.8Sr0.2Mn1-xCuxO3(x=0,0.1,0.2,0.3,0.4)比表面积Table 3 Specific surface area of La0.8Sr0.2Mn1-xCuxO3(x=0,0.1,0.2,0.3,0.4)tested by BET
为了探究催化剂表面元素及价态分布情况,采用X射线光电子能谱(XPS)进行分析,结果如图6所示。Mn 2p3/2轨道上拟合结合能641.5 eV和642.7 eV分别对应Mn3+、Mn4+物种。对于Cu 2p3/2轨道进行结合能拟合分峰,932.5 eV和934.6 eV信号峰分别属于Cu+、Cu2+物种。Cu+的出现主要来自于Mn4+/Mn3+和Cu2+/Cu+之间存在的相互作用[30]。对于O 1s轨道,528.9 eV和531.0 eV的信号峰分别来自于晶格氧(Olatt)和化学吸附氧(Oads)[31]。而化学吸附氧(Oads)更容易移动和脱附,因此在CO催化氧化中起主要作用。分峰拟合后的不同价态元素含量比如表4所示。Cu的掺杂会促进Mn4+/Mn3+比例升高,这表明催化剂还原能力得到增强,但掺杂量过大时,Mn4+所占比例下降,这表明金属离子价态变化可能会受到抑制,削弱了Cu和Mn之间的相互作用[18]。从表4可以看出,Cu+/Cu2+具有和Mn4+/Mn3+相似的变化趋势。
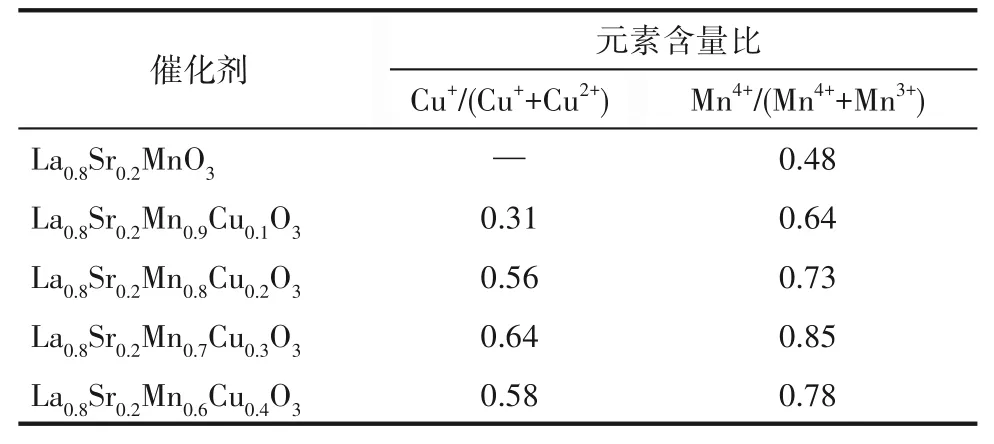
表4 La0.8Sr0.2Mn1-xCuxO3(x=0,0.1,0.2,0.3,0.4)催化剂表面不同价态元素含量比Table 4 Element valence state and relative content results of La0.8Sr0.2Mn1-xCuxO3(x=0,0.1,0.2,0.3,0.4)
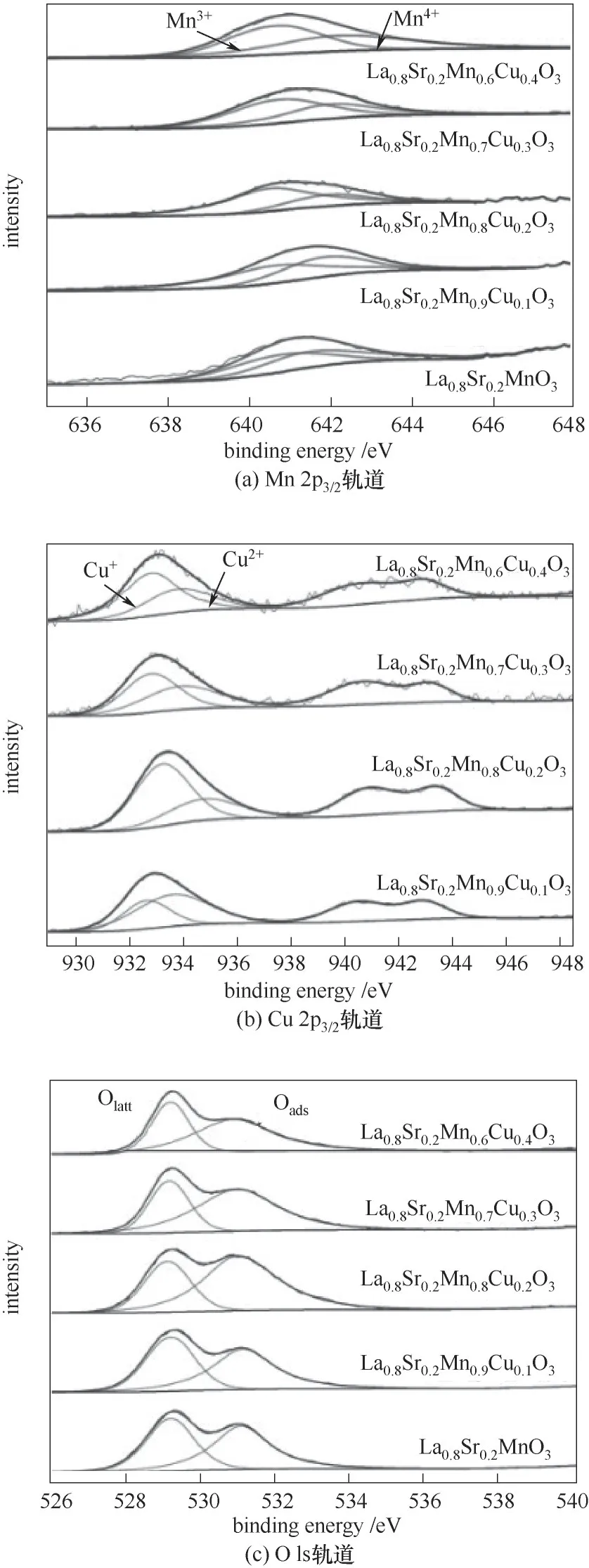
图6 La0.8Sr0.2Mn1-xCuxO3催化剂的XPS谱图Fig.6 XPS spectra of La0.8Sr0.2Mn1-xCuxO3 catalysts
La0.8Sr0.2Mn1-xCuxO3(x=0,0.1,0.2,0.3,0.4)催化剂的氧化还原性能采用H2程序升温还原(H2-TPR)分析,测试结果如图7所示。对于5种样品的H2-TPR曲线,均呈现出两个明显的信号峰,因此根据TPR峰值信号可将曲线分为低温区(<500℃)和高温区(>500℃)。结合此前XPS测试结果,Sr和Cu掺杂后,钙钛矿结构中Mn元素存在Mn4+和Mn3+两种状态。对于La0.8Sr0.2MnO3,低于500℃温度的低温区间主要发生氧空位化学吸附氧、活性晶格氧的还原,以及钙钛矿结构中Mn4+→Mn3+的还原[29]。同时,当催化剂中掺杂Cu2+后,低温区同时会进行Cu2+→Cu0的还原过程,与CuO直接还原不同的是,钙钛矿中Cu2+的还原需要以Cu+作为中间产物[32]。从XPS结果可知,适量的Cu取代,有助于提升Mn4+比例,能有效促进元素价态的转变,增强Cu和Mn的协同作用。高于500℃的高温区的还原信号主要与Mn3+还原为Mn2+有关。

图7 La0.8Sr0.2Mn1-xCuxO3(0≤x≤0.4)的H2-TPR曲线Fig.7 H2-TPR profiles of La0.8Sr0.2Mn1-xCuxO3(0≤x≤0.4)
从图中可以看出,与La0.8Sr0.2MnO3相比,Cu2+掺杂B位后,在低于500℃的温度区间内还原曲线具有明显差异。475℃左右发生的还原峰基本消失,在245℃附近出现新的还原峰,信号峰向温度更低的方向移动。还原温度降低的原因可能是由于Cu2+掺杂后,增强了氢溢流效应,H2分子受到活化,促进了H2分子和还原性物质的反应[33]。同时,Cu2+、Mn4+离子之间可能存在协同作用,对降低还原温度有一定影响。在低温区的还原峰强度随Cu2+掺杂量逐渐增强,这是因为Cu2+的引入会造成La0.8Sr0.2MnO3钙钛矿结构的扭曲对称度下降,因此更容易产生氧空缺位[34],钙钛矿的结构稳定性下降,更容易被还原。此外,由于Cu的掺杂量进一步增加,导致一些新的活性氧物种CuO和La2CuO4等出现并在钙钛矿催化剂表面富集,使还原峰信号进一步增强。随着反应温度进一步升高,在750℃附近出现新的还原信号,这主要发生钙钛矿结构中Mn3+→Mn2+的还原过程。信号峰对应的还原温度随Cu2+取代量的增加而逐渐降低,这可能是由于Cu2+还原过程中会释放大量的晶格氧,与催化剂产生更强的渗透晶格有关[32]。
由于H2-TPR表征中H2消耗量反映了催化剂还原能力的强弱及催化活性,对上述5种催化剂的H2消耗量进行定量分析,依次来比较Cu2+掺杂浓度对催化剂还原能力的影响。随着Cu2+掺杂,低温区(T<500℃)H2消 耗 分 别 为0.95、1.32、1.33、1.48、1.68 mmol/g,与La0.8Sr0.2MnO3相比,在引入Cu后,催化剂低温区还原峰的温度和强度都发生显著变化,这种降低意味着其氧化性总体得到增强。由于CO的催化氧化活性主要发生在低于500℃的低温区间,因此推测Cu2+的掺杂可能有助于催化剂对CO的催化氧化活性的提高[30]。5种催化剂的总氢气消耗量在2.30~2.50 mmol/g范围内,H2消耗量相差不大,La0.8Sr0.2Mn0.9Cu0.1O3具有最大的总氢气消耗量2.50 mmol/g。为进一步探究催化剂在低温区活性与催化剂比表面积之间的联系,对低温区H2消耗量与比表面积之间进行归一化处理,计算结果见表5。五种催化剂的归一化值较接近,催化剂活性的提高可能是由于较大的比表面积提高了活性中心的分散度,增加了反应物与活性中心接触的概率从而提高了催化性能[18]。
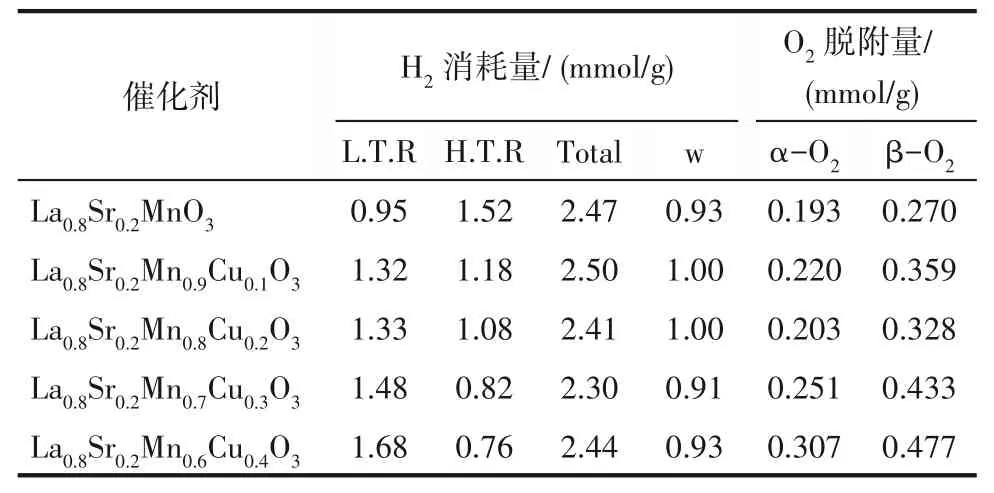
表5 La0.8Sr0.2Mn1-xCuxO3(x=0,0.1,0.2,0.3,0.4)的H2消耗量和O2脱附量Table 5 H2 consumption and O2 desorption of La0.8Sr0.2Mn1-xCuxO3(x=0,0.1,0.2,0.3,0.4)
为了探究Cu2+掺杂对催化剂氧物种的影响及氧物种浓度的影响,采用O2-TPD对钙钛矿催化剂进行表征,并进行定量计算。如图8所示,根据O2-TPD曲线特征,可将TPD信号峰划分为两个氧脱附温度区域[30]:小于550℃的脱附氧归因于催化剂表面化学吸附氧,是气态O2分子在催化剂表面缺陷位吸附,与氧空位有关,对催化剂的活性起主要作用,标记为α-O2;大于550℃的脱附氧主要来自钙钛矿结构中的晶格氧,与B位金属离子性质有关,标记为β-O2。从图中可以看出,5种催化剂的O2-TPD曲线具有相似的形状,在低于550℃温度范围内,La0.8Sr0.2MnO3的TPD信号强度不大且不存在明显的脱附峰,这说明其表面吸附氧数量不多。随着Cu2+掺杂比例增加,与La0.8Sr0.2MnO3相比,催化剂脱附峰强度开始增强,这说明Cu2+取代能促进催化剂产生更多的氧缺陷,表面氧空位数目得到增加,这对CO的催化氧化性能提升是有利的,这与之前研究报道一致[35]。随着反应温度进一步升高,值得注意的是,在600~700℃出现明显O2脱附峰,并随掺杂量的增加信号强度逐渐增强。这主要是钙钛矿中晶格氧的脱附,与Cu和Mn元素的还原有关,信号强度增强意味着晶格氧迁移率提高,Cu2+掺杂后使晶格氧更易从钙钛矿表面脱附。同时,Cu2+对Mn-O的键能起降低作用,促进钙钛矿结构中部分晶格氧活化并转移至表面从而脱附[36]。

图8 La0.8Sr0.2Mn1-xCuxO3(0≤x≤0.4)的O2-TPD曲线Fig.8 O2-TPD profiles of La0.8Sr0.2Mn1-xCuxO3(0≤x≤0.4)
对O2脱附曲线进行面积积分,分别计算α-O2和β-O2的脱附量,结果如表5所示。La0.8Sr0.2MnO3的α-O2和β-O2分别为0.193 mmol/g和0.270 mmol/g,在B位Cu2+部分取代后,钙钛矿的化学吸附氧和晶格氧数量都有所增加,并在x=0.4时达到最大的O2脱附量,分别为0.307 mmol/g和0.477 mmol/g。这表明Cu2+对B位金属离子的部分取代有助于提高催化剂化学吸附氧和晶格氧数量,同时结合BET测试结果,比表面积的增加也可能有助于O2吸附。
2.2 催化剂的催化活性
La0.8Sr0.2Mn1-xCuxO3(0≤x≤0.4)催化剂在CO催化氧化反应中的活性随温度的变化如图9所示。将CO催化转化率达到50%和90%时对应的温度值分别标记为T50和T90。从图9(a)可看出,所有催化剂的转化率曲线有着相似的形状,在温度升高后,催化活性逐渐提升。在首次CO催化氧化活性测试中,5种催化剂在120~160℃即可实现50%的CO转化率,在140~190℃即可实现90%的CO转化率,并随着温度的进一步提高,转化率增至100%。其中,La0.8Sr0.2Mn1-xCuxO3(0≤x≤0.4)在x=0.1时具有最优的CO催化活性,T50=119.4℃,T90=133.3℃,并在140℃达到100%的CO转化率。与La0.8Sr0.2MnO3相比,Cu2+掺杂后催化剂活性有较大提升,但催化活性随Cu2+取代比例呈现先增加后下降的趋势,但仍大于La0.8Sr0.2MnO3。这说明在A位Sr取代量为0.2时,由于少量Cu2+部分取代Mn3+会造成催化剂电荷不平衡,导致高价Mn4+产生,促进催化剂活性位点生成。同时,较小离子半径的Mn3+(0.0645 nm)被较大离子半径的Cu2+(0.0730 nm)部分取代后,钙钛矿晶格体积膨胀结构发生畸变,晶格缺陷的产生促进钙钛矿活性点位增加,有利于CO催化氧化活性的提高[18]。但随着Cu2+取代量的进一步增加,钙钛矿结构不对称度增大,结构稳定性下降,同时结合XRD及SEM可知,催化剂表面出现CuO和La2CuO4等氧化物,减少活性依附点位有效面积,从而削弱催化剂对CO的催化活性。
水蒸气和CO2的存在通常会影响催化剂活性,导致催化剂活性下降甚至失活。因此为探究火焰喷雾法制备的钙钛矿催化剂在水蒸气和CO2混合气体氛围中对CO的催化活性,模拟了实际含5%水蒸气和10% CO2的混合气体工况,测试结果如图9(b)所示。在混合气氛掺杂条件下,5种催化剂性能都有不同程度的衰减。Cu2+取代后的钙钛矿催化剂性能下降较La0.8Sr0.2MnO3明显,性能下降不超过15%。其中La0.8Sr0.2Mn0.9Cu0.1O3具有最高的CO催化活性,其T50和T90分别为128.8℃和150.2℃,对水蒸气和CO2具有良好的耐受性。Li等[37]研究了水蒸气对Cu/Mn基催化剂上甲苯催化氧化的影响,结果表明,随着水蒸气浓度的增加,甲苯反应可用活性点位减少,所有转化曲线向更高温度移动,催化活性下降。Yan等[38]对LaCo0.5Mn0.5O3催化剂的抗水中毒性进行了实验,在低于200℃时,水蒸气对催化剂活性点位的覆盖程度较高,水蒸气与CO在活性点位上形成竞争吸附。Deng等[39]发现在水蒸气和CO2共存时,Pt/CuCrOx催化剂对CO氧化性能受到抑制,并发现性能的下降归因于CO2占据活性点位并形成碳酸盐物种,减少了CO吸附从而导致催化剂失活。因此,推测La0.8Sr0.2Mn1-xCuxO3性能下降可能与表面活性点位较少有关,导致CO与活性中心接触概率下降,从而抑制CO的催化氧化[40]。
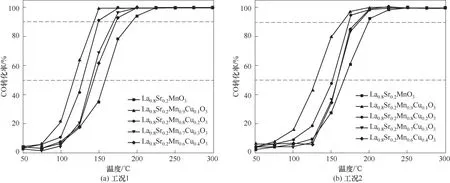
图9 La0.8Sr0.2Mn1-xCuxO3(x=0,0.1,0.2,0.3,0.4)在不同工况下的CO转化率Fig.9 Catalytic performance for CO oxidation of La0.8Sr0.2Mn1-xCuxO3(x=0,0.1,0.2,0.3,0.4)under different conditions
对两组工况的La0.8Sr0.2Mn1-xCuxO3催化剂分别进行5次连续催化氧化实验,以探究其催化性能的稳定性。测试结果如表6和表7所示,在循环测试中,每组催化剂性能随循环次数增加均有所下降,但降幅不超过10%。循环测试中,催化剂活性下降归因于CO扩散吸附于钙钛矿表面后,催化剂表面化学吸附氧和活性晶格氧参与氧化反应,氧空位和部分活性位点被逐渐消耗,导致催化剂活性有所下降。经历5次循环测试后,La0.8Sr0.2Mn0.9Cu0.1O3催化剂仍具有较优的催化性能,在工况1和工况2测试条件下 其T90分 别 为140.1℃和157.8℃。图10为La0.8Sr0.2Mn0.9Cu0.1O3在200℃恒定温度CO氧化反应长时间稳定性测试,可以看到该催化剂在40 h内始终保持较高的CO催化转化率,在加入水蒸气和CO2后催化剂仍保持优越的稳定性和良好的抗烧结性。表8列举了近年制备钙钛矿催化剂与火焰喷雾法制备催化剂对CO氧化的催化性能比较,火焰喷雾法制备的催化剂具有良好的CO催化活性。结合上述微观表征及CO催化氧化实验,火焰喷雾法制备的La0.8Sr0.2Mn1-xCuxO3催化剂表现出良好的催化活性和抗烧结能力,适合于催化氧化CO。
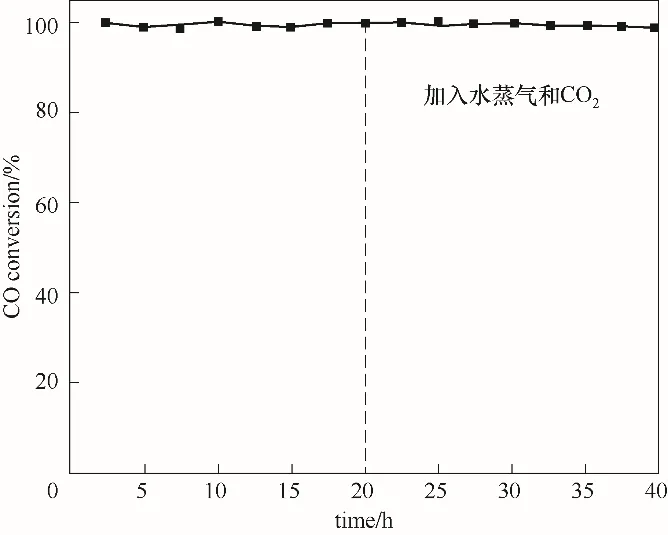
图10 La0.8Sr0.2Mn0.9Cu0.1O3催化剂的CO氧化反应稳定性(反应温度200℃)Fig.10 Stability of La0.8Sr0.2Mn0.9Cu0.1O3 catalyst at 200℃for CO oxidation

表6 工况1连续催化氧化实验中CO转化率为50%和90%时的对应温度Table 6 Temperature of 50%and 90%conversion in consecutive CO catalytic oxidation of case 1

表7 工况2连续催化氧化实验中CO转化率为50%和90%时的对应温度Table 7 Temperature of 50%and 90%conversion in consecutive CO catalytic oxidation of case 2
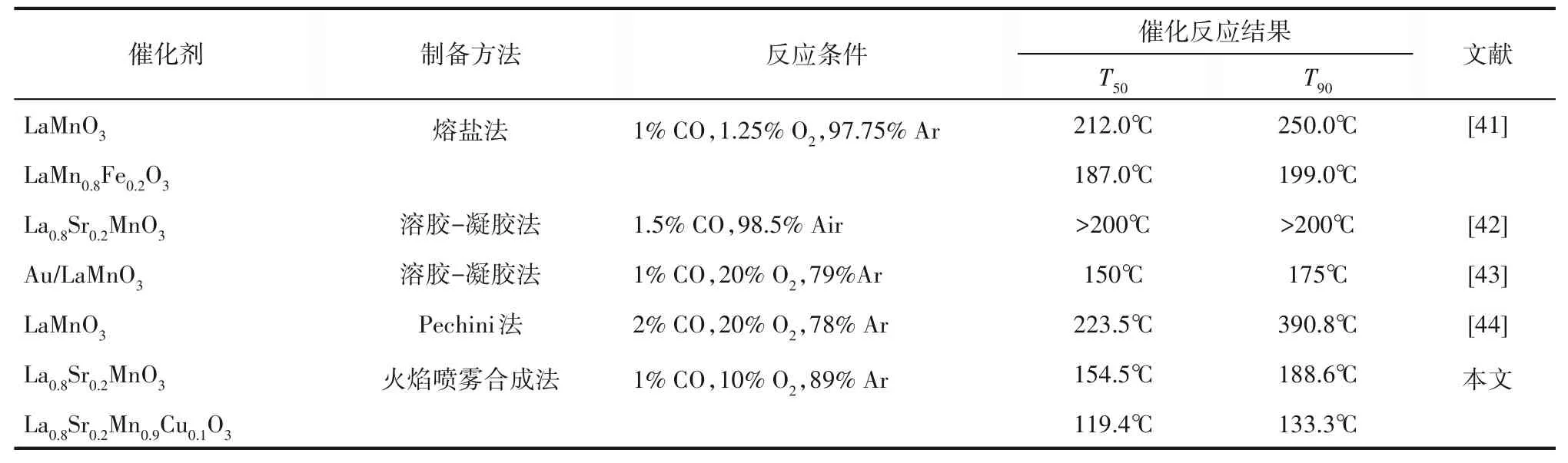
表8 文献中具有代表性钙钛矿催化剂与本文La0.8Sr0.2Mn1-xCuxO3(0≤x<1)催化剂对CO氧化的催化性能比较Table 8 Comparison of La0.8Sr0.2Mn1-xCuxO3(0≤x<1)catalyst for CO oxidation between references and this work
3 结 论
本文采用火焰喷雾合成法成功制备了用于CO催化氧化的La0.8Sr0.2Mn1-xCuxO3(0≤x≤0.4)钙钛矿催化剂。在Sr2+取代量为0.2时,少量Cu2+掺杂仍能保持良好的钙钛矿相,但浓度增加后会出现杂相偏析,Cu2+脱离钙钛矿晶格结构,形成CuO和La2CuO4等氧化物。这也表明钙钛矿结构稳定性下降,不利于CO催化氧化。催化剂样品具有较精确的化学成分组成和良好的疏松多孔结构,其比表面积在9.85~17.68 m2/g范围内,并随Cu2+的掺杂,比表面积逐渐增大。由H2-TPR测试结果可知,Cu2+部分取代后,催化剂低温区域的催化活性显著增强,这是由于Cu2+和Mn4+之间存在协同作用,共同促进催化活性提升。O2-TPD结果显示,随着Cu2+取代量的增加,晶格氧迁移率提高,Cu2+掺杂后使晶格氧更易从钙钛矿表面脱附。
在CO催化氧化测试中,5种催化剂均分别在160℃和190℃以下即可实现50%和90%的CO转化率,并随着温度的升高,CO完全氧化转化。其中,La0.8Sr0.2Mn0.9Cu0.1O3具有最优的CO催化活性,其T50和T90分别为119.4℃和133.3℃,并在140℃达到100%的CO转化率。研究了火焰喷雾合成法制备的钙钛矿催化剂在水蒸气和CO2混合气体氛围下的催化活性,测试结果表明,5种催化剂都有不同程度的性能衰减,但性能最优的La0.8Sr0.2Mn0.9Cu0.1O3钙钛矿催化剂T90=150.2℃。催化氧化活性下降可能是CO2和水蒸气与CO在催化剂表面形成竞争吸附,占据催化剂表面活性点位,从而抑制CO的催化氧化。值得注意的是,在连续稳定性催化氧化测试中,5种催化剂性能衰减不超过10%,这表明火焰喷雾合成法制备的钙钛矿催化剂具有良好的稳定性和抗烧结能力。综上所述,火焰喷雾合成法十分有利于制备高CO氧化活性的钙钛矿催化剂。
