基于TCGA数据库构建前列腺癌相关miRNA预后模型
2021-10-13张泉波李玉莲陈绮张艳艳陈国荣
张泉波 李玉莲 陈绮 张艳艳 陈国荣
前列腺癌是男性最常见的泌尿系统恶性肿瘤之一,全世界每年新发病例约130万例[1-3]。尽管前列腺癌的治疗策略已有了明显进步,患者的存活率得到了显著提高[4],但患者的总体生存期仍未得到提升,5年总存活率只有28%左右[5-6]。目前,患者的预后评估方法主要依靠临床TNM分期和常规组织病理学检查[7-8],然而这并不能准确评价前列腺癌患者的预后情况,因此迫切需要新的早期诊断标志物以及治疗靶点来协助改善前列腺癌患者的预后。miRNA是一类短链非编码RNA,目前科学家已经发现了1 400多个人类miRNA,这些miRNA可以通过抑制翻译或促进mRNA降解从而调节基因的表达[9-10]。众所周知,miRNA可以通过调节靶基因的表达来影响多种生物途径,如细胞增殖、细胞分化、血管生成、肿瘤侵袭迁移等[11-12]。Wei等[13]研究发现miR-296可以通过减少高迁移率族蛋白A1(HMGA1)基因的表达来抑制前列腺癌细胞的增殖与侵袭。Gan等[14]研究证明miR-16-5p能够调节相关通路导致细胞停滞在G0/G1期,从而提高前列腺癌细胞对放射治疗的灵敏度。这些研究均表明,许多miRNA或许可以作为前列腺癌的诊断标志物或治疗靶点。虽然已有研究证实了个别miRNA与前列腺癌预后之间的关系[15-17],但目前仍没有研究能够系统地阐明前列腺癌中的差异表达miRNA与预后之间的关联。因此,本研究利用癌症基因组图谱数据库(the cancer genome atlas,TCGA)中前列腺癌患者的miRNA和mRNA数据及临床信息,构建由miRNA组成的预后风险评分模型,为后续研究提供理论基础。
1 材料和方法
1.1 基因数据的提取 从TCGA数据库(https://portal.gdc.cancer.gov/)中下载了499例前列腺癌组织样本和52例正常组织样本的miRNA和mRNA数据,并确立筛选标准:(1)错误发现率(false discovery rate,FDR)<0.05;(2)表达差异>2 倍以上[log2差异倍数(fold change,FC)>1,FC>2]。最终筛选出131个差异表达miRNA及524个差异mRNA,利用R语言绘制了火山图和热图。
1.2 Kaplan-Meier预后分析 分别利用R语言的survival包以及基因表达谱动态分析(gene expression profiling interactive analysis,GEPIA)(http://gepia2.cancerpku.cn)数据库对前列腺癌患者的差异表达miRNA及mRNA进行预后分析。
1.3 构建miRNA-mRNA调控网络 利用TargetScan(http://www.targetscan.org)、miRDB(http://mirdb.org/miRDB)和miRanda(http://www.microrna.org)数据库对纳入模型的miRNA进行靶基因预测。使用 Venny 2.1(http://bioinfogp.cnb.csic.es/tools/venny/index.html)来进一步确认这些靶基因的生物信息学可信度。使用Cytoscape软件绘制miRNA-mRNA调控机制网络。
1.4 基因功能富集分析 基因本体论(gene ontology,GO)的功能注释由生物学过程(biological process,BP)、细胞组分(cellular component,CC)和分子功能(molecular function,MF)3部分组成,通过京都基因和基因组百科全书(Kyoto encyclopedia of genes and genomes,KEGG)数据库分析相关信号通路。
1.5 统计学处理 差异miRNA及mRNA表达比较采用两独立样本t检验;不同临床病理分期的基因表达水平比较采用单因素方差分析;基因表达与患者预后的关系分析采用Kaplan-Meier生存曲线,两组生存率比较采用log-rank检验。P<0.05为差异有统计学意义。
2 结果
2.1 前列腺癌差异表达miRNA筛选及预后鉴定 按照预先设定的筛选标准对所获取的miRNA数据进行差异分析并进一步进行Kaplan-Meier生存分析探究预后相关的miRNA,结果提示miR-19a-3p、miR-144-3p、miR-223-5p和miR-483-3p与患者总生存期(overall survival,OS)密切相关,miR-144-3p、miR-223-5p、miR-483-3p高表达组OS均高于低表达组(均P<0.05),而miR-19a-3p高表达组OS低于低表达组(P<0.05)[图1(见插页)和图2]。

图1 差异表达miRNA(a:前列腺癌组织与正常组织之间的差异miRNA基因热图;b:差异miRNA火山图,红点代表上调的miRNA,绿点代表下调的miRNA)

图2 差异表达miRNA与患者总生存期的关系
2.2 miRNA表达量与前列腺癌患者的临床病理特征的关系 4个预后相关miRNA中,miR-483-3p表达量与T分期有关,在不同T分期患者中呈现为T4>T2>T3(P<0.05);miR-19a-3p表达量与患者年龄有关,>60岁以上患者miR-19a-3p表达量明显高于≤60岁患者(P<0.05);miR-223-5p表达量与Gleason评分有关,在7分时表达量最高,而≤6分时表达量最低(P<0.05)(图3)。

图3 miRNA表达量与前列腺癌患者的临床病理特征的关系(*P<0.05,**P<0.01)
2.3 预测相关靶基因 利用TargetScan、miRDB和mi-Randa数据库预测4个miRNA作用的靶基因,结果显示 miR-19a-3p、miR-144-3p、miR-223-5p 和 miR-483-3p潜在靶基因分别有713、521、420和49个(图4)。随后结合TCGA数据库中mRNA差异分析结果,发现在前列腺癌中上述4个miRNA的潜在靶基因有41个(图5,见插页)。既往已有文献报道,miRNA可以通过结合mRNA导致基因沉默[18],因此最终确定4个miR-NA的潜在靶基因有33个并构建miRNA-mRNA调控网络显示其关系(图6)。

图4 差异miRNA的靶基因预测
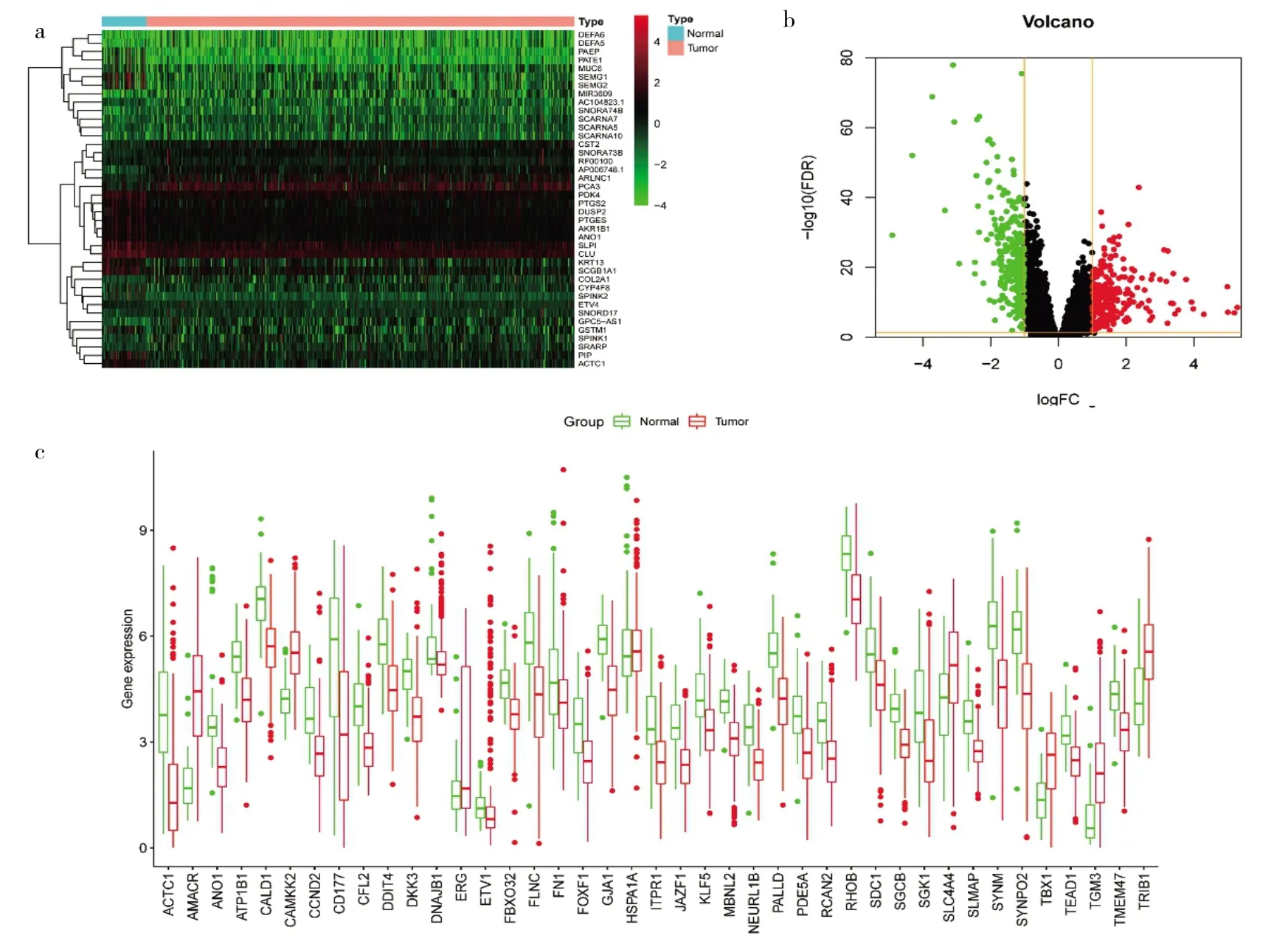
图5 差异表达mRNA(a:前列腺癌组织与正常组织之间差异mRNA基因热图;b:差异mRNA火山图,红点代表上调的基因,绿点代表下调的基因;c:41个差异mRNA的表达量)
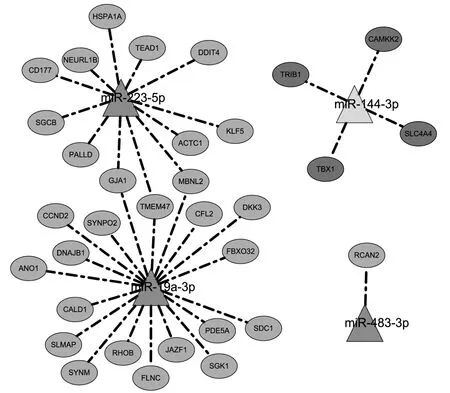
图6 miRNA与靶基因调控网络(深灰色三角形代表上调的miRNA,浅色三角形代表向下调节的miRNA;浅灰色椭圆代表下调的mRNA,而深灰色椭圆代表上调的mRNA)
2.4 差异表达基因的功能富集分析 对这33个潜在靶基因进行GO功能富集分析,表明其BP主要与肌肉细胞分化、发育以及上皮细胞发育有关;CC主要与肌原纤维、纤维的收缩和细胞-基质黏附连接相关;MF主要与肌动蛋白结合和细胞黏附分子结合相关。此外,KEGG通路分析结果提示靶基因主要涉及到叉头转录因子(forkhead box sub-group O,FoxO)和磷脂酰肌醇 3激酶/蛋白激酶B(phosphatidylinositol 3 kinase/AKT serine/threonine kinase 1,PI3K/Akt)信号通路(图7,见插页)。
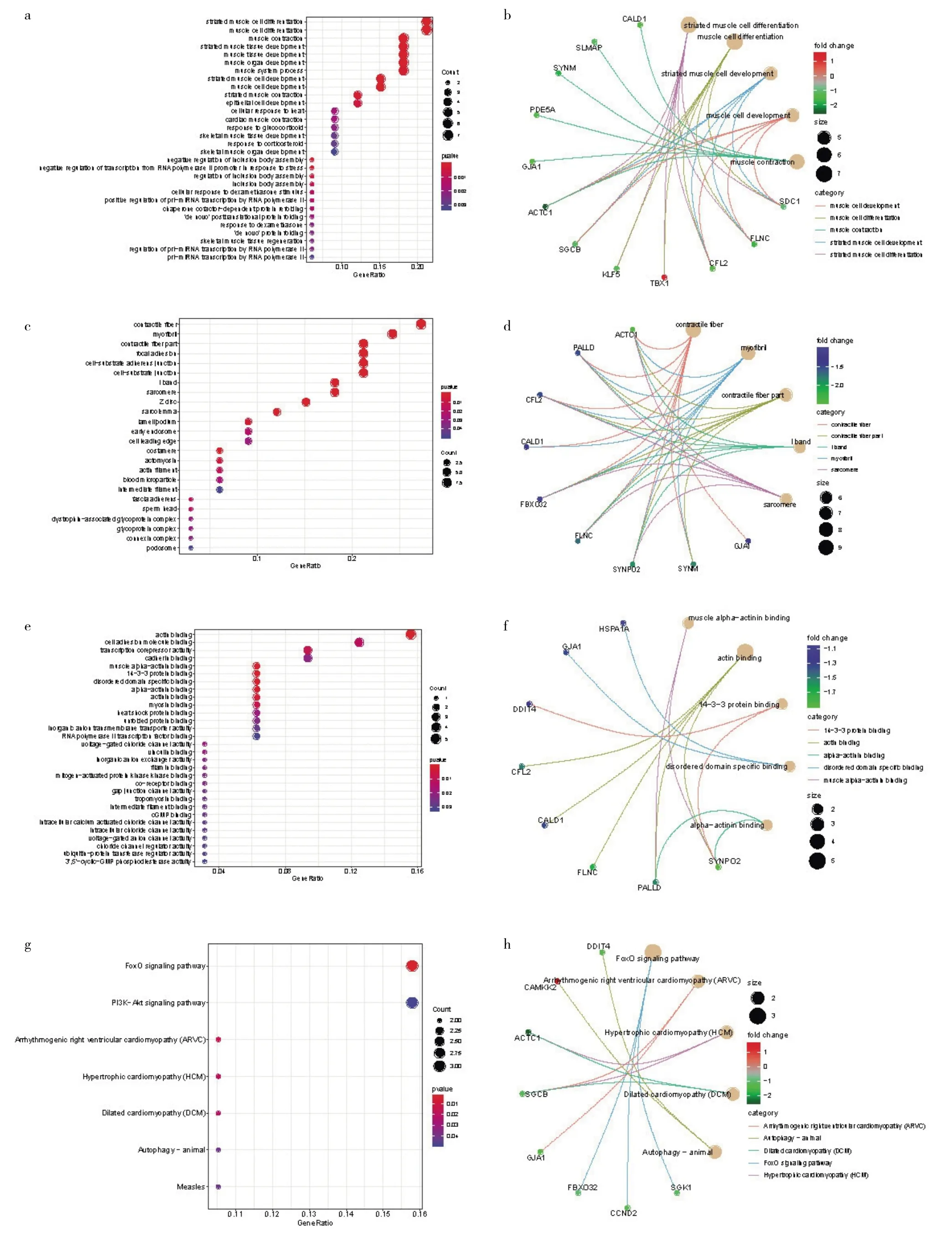
图7 差异表达基因的功能富集分析[a-b:生物学过程(BP);c-d:细胞组分(CC);e-f:分子功能(MF);g-h:京都基因和基因组百科全书(KEGG)通路分析]
2.5 差异表达基因与患者预后的关系 Kaplan-Meier生存分析显示,前列腺癌患者心肌肌动蛋白α1(actin alpha cardiac muscle 1,ACTC1)、CD177、丝切蛋白 2(cofilin 2,CFL2)、肌蛋白修饰调节因子2(muscleblind like splicing regulator 2,MBNL2)、含钯蛋白(palladin,PALLD)、磷酸二酯酶 5A(phosphodies terase 5A,PDE5A)、联丝蛋白(synemin,SYNM)或突触极蛋白 2(synaptopodin 2,SYNPO2)高表达组患者无病生存期均长于低表达组患者(均P<0.05)(图 8)。

图8 差异表达基因与患者预后的关系(ACTC1为心肌肌动蛋白α1;CFL2为丝切蛋白2;MBNL2为肌蛋白修饰调节因子2;PALLD为含钯蛋白;PDE5A为磷酸二酯酶5A;SYNM为联丝蛋白;SYNPO2为突触极蛋白2)
3 讨论
随着对前列腺癌研究的日益深入,患者的预后得到了极大的改善。然而,由于对前列腺癌的筛查指标仍有争议,因此,进一步探索前列腺癌的相关机制,挖掘有效的基因靶点对前列腺癌的诊断治疗及预后评估具有重要的临床意义[19-20]。
越来越多的研究表明miRNA可以建立复杂的基因调控网络,并参与多种生物途径,从而影响癌症的发病机制[21-22]。在肿瘤发生、发展中,miRNA是一把双刃剑,肿瘤类型、临床阶段、遗传背景,甚至治疗方案都能影响到它在肿瘤中的作用[23]。
本研究所构建的预后模型共纳入4个miRNA,分别为 miR-19a-3p、miR-144-3p、miR-223-5p 和 miR-483-3p。miR-19a-3p已被证实在乳腺癌、肝细胞癌及胶质瘤等[24-25]多种癌症中呈现为低表达状态,而其在前列癌细胞系中的表达量却大不相同,如miR-19a-3p在原发性前列腺癌细胞22RV1中表达量上升,但在骨转移前列腺癌细胞系(PC-3、C4-2B和VCAP)中明显减少。现有研究表明,miR-19a-3p可以通过调节TGF-β通路的下游SMAD家族成员2(SMAD2)和SMAD家族成员4(SMAD4),从而影响前列腺癌细胞的迁移和侵袭[26]。此外,miR-144-3p被证实参与了多种恶性肿瘤的发生、发展,相关研究显示它可以作为肿瘤抑制因子,通过下调中心体蛋白55(CEP55)从而诱导前列腺癌细胞的凋亡,并通过调节细胞周期来抑制细胞增殖[27]。miR-483-3p与多种恶性肿瘤的发生、发展具有密切的关联。研究表明,miR-483-3p可以通过与p53凋亡上调基因(PUMA)mRNA 3'UTR相互作用,从而抑制抗凋亡因子B细胞淋巴瘤因子3(BCL3)和B细胞淋巴瘤因子XL(BCLXL)来发挥抗凋亡作用[28]。此外,miR-223-5p与乳腺癌、胃癌和肝细胞肝癌等多种肿瘤细胞的侵袭和迁移相关。有研究表明,miR-223-5p在非小细胞肺癌组织中呈低表达趋势,研究人员在体内外实验中上调了miR-223-5p的表达量后,发现E2F转录因子8(E2F8)作为miR-223-5p的功能靶点,出现了表达量的下降,并抑制非小细胞肺癌细胞的增殖、迁移和侵袭[29]。虽然已有研究证明miR-483-3p、miR-223-5p与肿瘤之间的相关性,但是暂时没有研究能证明这两种miRNA与前列腺癌患者预后的关联。
为了探索 miR-19a-3p、miR-144-3p、miR-223-5p和miR-483-3p的潜在靶基因,本研究通过数据库及基因功能富集分析,最终证实了ACTC1、CD177、CFL2、MBNL2、PALLD、PDE5A、SYNM、SYNPO2 均与前列腺癌患者预后相关。ACTC1作为肌动蛋白家族中的一员,早期研究发现其在心力衰竭、高血压等心血管疾病中起到重要作用;而在后续的研究中,发现其在肿瘤的发生、发展中也发挥着关键作用;现有研究发现,前列腺癌雄激素剥夺治疗后患者中ACTC1基因表达量上调[30]。CD177作为一种糖磷脂素基醇-细胞外膜结合蛋白,属于Ly-6家族[31]。研究表明CD177的下调与β-连环蛋白(β-catenin)信号传导的增加有关,它能够作为上皮细胞增殖的调节因子参与前列腺癌的进展[32]。CFL2基因是肌动蛋白结合蛋白家族的重要成员,主要在哺乳动物的骨骼肌和心肌中表达,对维持肌肉正常的生长发育和功能起着非常重要的作用。研究发现CFL2基因可通过miR-1299/CFL2、miR-369-3p/CFL2等多种途径影响前列腺肿瘤的进展[33-34]。研究发现PDE5A的抑制剂可以通过放大有丝分裂阻滞信号,协同长春新碱在去势抵抗性前列腺癌中起到治疗作用[35]。SYNPO2作为一种肌动蛋白结合蛋白和侵袭性癌症生物标志物,在细胞体内诱导形成复杂的应激纤维网络。已有研究表明SYNPO2可以通过诱导周围肌动蛋白束的组装从而促进PC-3前列腺癌细胞的迁移[36]。虽然已有研究证明了MBNL2、PALLD、SYNM与肿瘤之间的相关性,但是暂时未能证明这两种miRNA与前列腺癌患者预后的关联[37-39]。
综上所述,本研究对miRNA数据进行了综合分析,为探索差异表达miRNA在前列腺癌中的作用提供了有效的统计方法,然而本研究也存在一定的局限性。首先,本研究的数据仅来源于TCGA数据库,应该结合更多数据库来增加样本量;其次,所筛选到的 mRNA表达与生存情况未在蛋白质层面进行分析,未对有研究价值的调控关系进行功能实验验证,这些将在后续研究中进一步完善。
