基于第一性原理计算L12-Al3Li金属间化合物低指数晶面表面能
2021-09-03房洪杰姚建刚
张 倩, 王 营, 房洪杰, 余 琨, 2, 江 勇, 2, 姚建刚
(1. 烟台南山学院 材料科学与工程学院, 山东 烟台 265713;2. 中南大学 a. 材料科学与工程学院, b. 教育部有色金属材料重点实验室, 湖南 长沙 410083)
金属间化合物L12-Al3Li(δ′-Al3Li)是Al-Li合金的重要析出相,对改善Al-Li合金的强度和高温抗蠕变性能具有重要的作用。近几十年来,Al-Li合金的基本热力学性能一直是铝合金领域的研究热点之一[1-5]。Al-Li 合金的高弹性模量与Al-Al3Li界面特征密切相关[6],因此,研究L12-Al3Li与基体Al之间的界面性质具有重要意义。
Baumann等[7]、 Mao等[8]分别根据理论和实验计算了Al-Al3Li的界面能。高英俊等[9-10]从原子成键的角度,讨论了Al3Li相的价电子结构,并且运用固体经验电子理论的电子密度分析法,对比计算δ-Al3Li 和δ′-Al3Li相与基体之间的界面电子性质的差异,认为 Al-δ′-Al3Li的界面电子密度在较小的应力下保持连续,说明δ′-Al3Li相与基体结合良好,起到了增强界面强度的作用。
析出相的表面性质是研究金属材料界面特征的基础。 虽然对于Al晶格的表面性质的理论和实验研究有很多报道[11-13], 但是Al-Li合金析出相的表面特性研究较少, 特别是 Al3Li表面性能的研究至今未见报道。 本文中从理论上计算 L12-Al3Li金属间化合物(100)、 (110)、 (111)3个低指数晶面的结构和表面能, 以期为相关实验研究提供参考。
1 计算方法
本文中所有计算都在基于密度泛函理论的原子尺度材料模拟的计算机软件包VASP[14]中进行。 选择投影缀加平面波赝势(PAW)[15]描述离子-电子间的相互作用, 采用广义梯度近似(GGA)中的Perdew-Burke-Ernzerhof(PBE)交互关联泛函法[16]处理电子间的交互关联作用。 在计算Al、 Li、 Al3Li单胞模型以及Al3Li金属间化合物(100)、 (110)、 (111)3个低指数面晶面时, 简约布里渊区的K点网格(尺寸为16×16×16)采用Monkhorst-Pack方法来划分。所有单胞及表面波函数动能截断能取值为320 eV。 驰豫后的能量和原子受力的收敛判据分别为1×10-4eV和 0.2 eV/nm。
2 结果与讨论
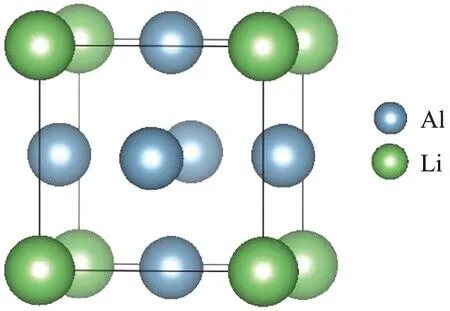
图1 L12-Al3Li金属间化合物的晶胞结构
对Al、 Li及L12-Al3Li单胞进行包括体积和原子坐标在内的几何优化。 通过Birch-Murnaghan状态方程拟合方法得到Al、 Li及L12-Al3Li的晶格常数、体模量、基态能,结果见表1。由表可以看出,计算结果与实验值接近,表明本文中采用的计算参数是合理的。

表1 Al、 Li和L12-Al3Li的晶格常数、体模量、基态能的计算值和实验值
2.1 L12-Al3Li金属间化合物的表面能计算
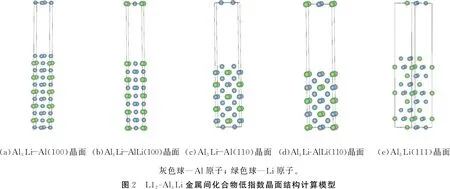
在计算L12-Al3Li金属间化合物(100)、 (110)、 (111)3个低指数晶面的表面能之前,首先要确定相应晶面的表面终端。运用材料性能模拟(MS)软件及晶体结构可视化软件(VESTA),可以判断L12-Al3Li金属间化合物的(111)晶面只有一种终止方式,即符合Al3Li化学计量比的表面终端。(100)、(110)晶面具有Al-Li表面终端以及富Al表面终端2种模式,在进行相关计算时,对所有表面类型全部予以考虑。
本文中计算表面能采用的是添加真空层的界面模型, 并用三维周期性边界条件的层晶和超胞模型进行模拟。 为了避免表面法向上原子间产生相互作用而影响计算精度, 通常要在表面模型中增加足够厚的真空层。 真空层根据体系大小确定, 经过逐渐加厚测试计算, 本文中采取厚度为12 Å的真空层即可以保证计算的准确性。 在构建表面模型时, 不同的表面层数对计算结果影响是不同的。 当层数过少时, 层与层之间的相互作用对表面能的结果会产生影响; 当层数过多时,不仅会增加计算量, 而且表面模型中块体Al3Li的成分不断增大, 导致块体能量在表面能中的比例增大, 表面模型失效,计算值不能反映表面能的真实值。构建表面模型还应该使表面模型具有中心对称性, 这样才能根据定义给出表面能计算公式。 同时, 具有中心对称的结构可以抵消因上、 下表面电偶极矩存在而带来的表面能计算误差。
综合考虑各种因素,分别对(100)、(110)、(111)晶面Al-Li 终端的表面模型构建9层表面超胞模型,而(100)、(110)晶面富Al终端构建11层表面超胞模型。在结构驰豫时,采取固定中间层,上、下4(或5)层同时驰豫的方法。L12-Al3Li金属间化合物(100)、(110)、(111)3个低指数晶面面的表面结构模型如图2所示。
表面能是反映表面稳定性的重要参数。根据定义,具有中心对称结构的 L12-Al3Li金属间化合物晶体表面模型的表面能σ计算公式为
(1)
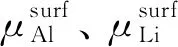
很显然,Al原子和Li原子在表面中的化学势要高于在体相中的化学势,形成块体的Al3Li时,应满足以下方程:
(2)
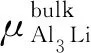
根据式(1)、(2)可得如下方程:
(3)
对于化学计量比表面, 3NLi=NAl,因此表面能公式可改写为
(4)
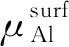
为了回避上述问题,Wang等[19]给出一个可行的计算方案。对于同一个非化学计量比表面,如果存在2种表面终端,那么其表面能可用两者的平均值来确定。鉴于此,L12-Al3Li金属间化合物的(100)、 (110)晶面的表面能即可取Al-Li表面终端((Al-Li)ter)的表面能和富Al表面终端((Al)ter)的表面能的平均值,显然,(3NLi-NAl)(Al-Li)ter=-(3NLi-NAl)(Al)ter。
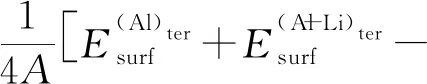
(5)
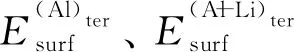
根据式(4)、(5)计算得到L12-Al3Li金属间化合物(100)、 (110)、 (111)3个低指数晶面的表面能,如表2所示,同时给出不同物质相应晶面的表面能数据,以比较计算结果的准确性。
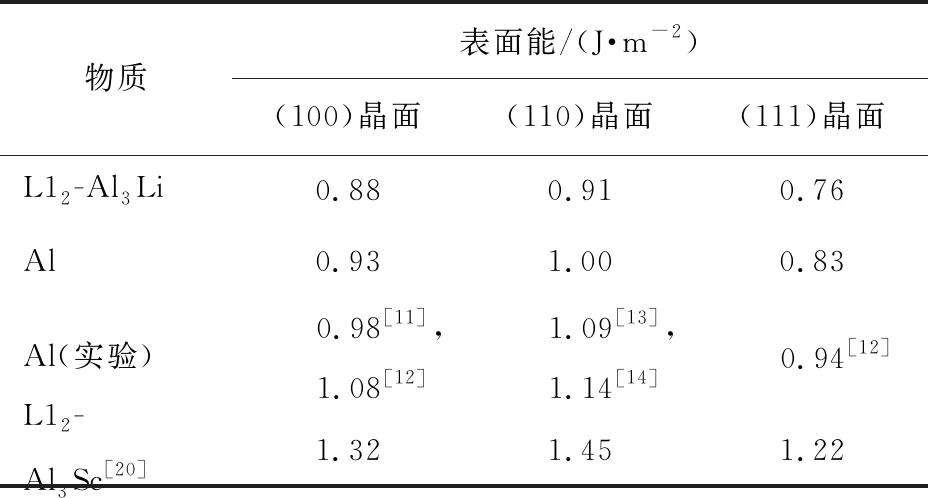
表2 不同物质的低指数表面能的计算值和Al原子的实验值
计算结果表明, 在L12-Al3Li金属间化合物的3个低指数晶面中, (111)晶面的表面能最小, 约为0.757 J/m2, (110)晶面的表面能最大, (100)晶面的居中。 表面能的本质是反映表面原子悬挂键数量的多少。 不同指数晶面的原子排布方式不同,原子密度也不同, 通常来说, 密排面原子排布最紧密, 相应的表面原子悬挂键数量最少, 能量最低。 因为(111)晶面正好为密排面, 所以其能量最低, 稳定性也最好。
由于缺乏相应的实验数据, 因此, 本文中用同样方法给出了同为面心立方结构Al的3个低指数面的表面能(见表2)进行比较。 从表中可以看出, 表面能计算值与实验值吻合很好,说明所用方法是可信的。 此外, 表2中还列出Sun等[20]从理论上给出的L12-Al3Sc金属间化合物的3个低指数晶面的表面能数值。 通过比较可以看出, 该金属间化合物的表面能与L12-Al3Li金属间化合物、 Al的变化规律相似, 都是(111)面最稳定。 另外还发现, 与Al及L12-Al3Sc金属间化合物相比, L12-Al3Li金属间化合物的3个低指数晶面的表面能是最小的, 这应该与Li原子的原子半径有关。 由于与Al、 Sc原子相比, Li原子不仅具有最大的原子半径, 而且最外层电子排布为 1s22s1, s轨道电子云为球形分布, 在各个方向分布的概率是一样的, 因此Li原子电子云在空间更加弥散, 更容易与附近其他原子成键, 从而减少表面原子悬挂键的数目, 使相应的晶面具有更好的稳定性。
2.2 L12-Al3Li金属间化合物的表面电子性质
为了形象地反映表面能的大小,本文中同时计算了L12-Al3Li金属间化合物3个低指数晶面超胞的电子局域函数(ELF),结果见图3。通常来说,ELF数值越大,表明原子间的电子聚集越多。由图可以看出,3个超胞内部ELF的值约为0.5,为典型的金属键所具有的“电子海”特征,即Al原子与Li原子、Al原子与Al原子间金属键起主导作用。在所有超胞表面原子的上方,ELF的值明显增大,表面电子数聚集产生了有大量的未配对电子,表面能越大,电子数目越多。其中(100)-AlLi和(110)-AlLi这2个超胞表面原子的局域性最强,而(111)晶面为化学计量比表面,表面原子的电子分布与内部原子较为接近,电子悬挂键最少,因此具有最小的表面能。这与上一节中的计算结果是一致的。
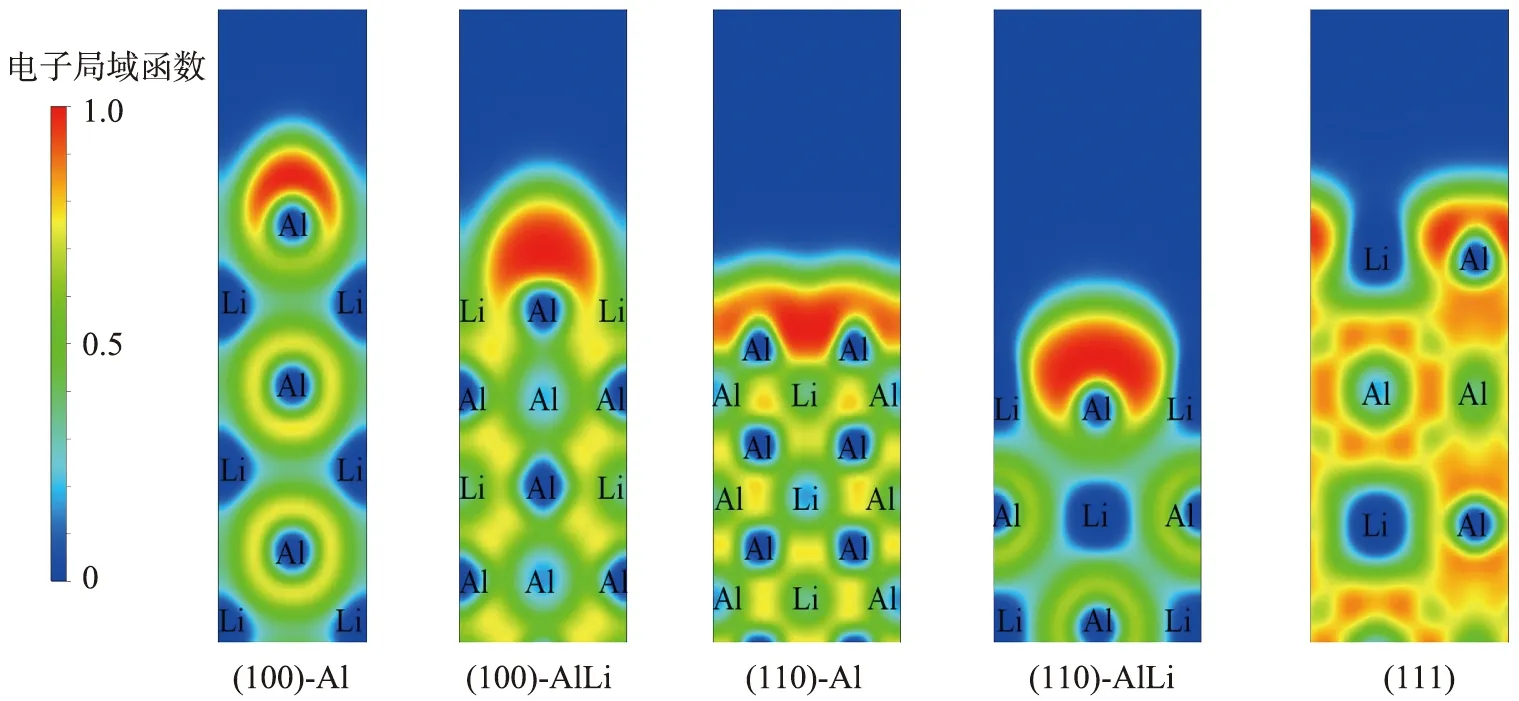
图3 L12-Al3Li金属间化合物低指数晶面的电子局域函数
3 结论
L12-Al3Li金属间化合物的(100)、 (110)晶面存在Al-Li表面终端以及富Al表面终端2种非化学计量比模式,因此两者的表面能大小与Al原子的化学势有关。(111)晶面只有一种化学计量比表面终端结构,其表面能的大小与Al原子化学势无关。L12-Al3Li金属间化合物的3个低指数表面中,(111)晶面为原子密排面,表面能最小,最稳定;(100)晶面居中,(110)晶面的表面能最大。
