基于深度学习和支持向量机的基因结合蛋白预测
2021-09-03陈佐瓒丁小军甘井中
陈佐瓒, 徐 兵, 丁小军, 甘井中
(1. 玉林师范学院 计算机科学与工程学院, 广西 玉林 537000; 2. 南京师范大学 地理科学学院, 江苏 南京 210023;3. 中南大学 计算机学院, 湖南 长沙 410083)
基因与蛋白质的结合是生物体的重要功能。随着科技的进步,基因测序技术不断完善,诸多专家学者致力于挖掘基因序列,探索蛋白质在生物学上的意义[1-3]。每个生物都有蛋白质,预测蛋白质原始序列是当今生物信息学领域的研究热点[4-6]。蛋白质与脱氧核糖核酸(DNA)结合的能力称为DNA结合蛋白(DBP)。 DNA通过与蛋白质结合,可以实现多种功能, 调节生物体的机制[7]。 生物信息学领域的热点问题集中在计算机资源和一些分类算法的集成上[8-9],其中蛋白质数据的积累、人工计算工作量以及人力物力成本等都是需要考虑的问题。
近年来,一些学者认为DNA结合蛋白预测是一个分类任务,因此诸多基于统计学和机器学习方法应用于DNA结合蛋白研究[10-13]。以上方法确实比人工分类方法的效率有所提升,但是在预测精度和速度方面还需要提升和改进。基于统计学的生物实验预测方法的优点是预测效果好,准确性极高,但也存在成本高、预测时间长的缺点。基于机器学习算法通过蛋白质的结构以及功能特性来学习其特征集合,采用机器学习中非线性映射方法,根据集合特征实现分类,但是如何保持集合向量分类,获得可以有效输出特征分类的结果还需要重点研究[14]。目前,人工智能中的深度学习方法已成为DNA结合蛋白预测方向上生物学信息的研究热点, 并取得了显著成果[15],但是,在当今日益增长的生物数据中,如何使用当前的深度模型来解释生物信息甚至生物问题,是一个很有意义的研究课题。基于此,本文中提出一种基于深度学习和支持向量机(SVM)的DNA结合蛋白预测算法(简称本文算法)。
1 DNA结合蛋白预测方法
1.1 模型框架
给定结构序列A1A2A3A4A5A6A7…AL,该结构序列包含20个碱性氨基酸和噪声蛋白,长度为L。通过嵌入操作,采用卷积神经网络(convolutional neural network,CNN)和门控循环单元 (gated recurrent unit, GRU)深度学习方法构建模型(见图1),构建深层神经网络对原始氨基酸序列进行编码和解码,得到氨基酸序列预测结果。通过特定的氨基酸序列的预测实例,分析模型各个模块的功能。
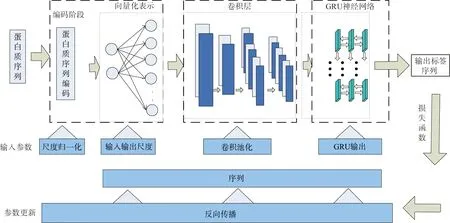
GRU—门控循环单元。
在图1所示的模型框架结构中,输入流为模拟氨基酸序列“MSFMVPT”特征的工作流程,主要包括4个阶段: 1)原始氨基酸序列成为固定长度的整数序列,需要进行编码; 2)通过嵌入操作将特征序列进行向量化表示; 3)将经过编码标注后的特征序列转化得到的特征向量馈入Convolution(卷积)中,进行特征提取; 4)不同于传统卷积神经网络常用的Softmax分类器方法,本文中将提取后的特征序列馈入到GRU中进行解码输出,该输出为一个定长的向量,然后通过SVM进行分类输出。
1.2 序列编码
本文算法的一个显著特点是需要对原始的蛋白质序列进行馈入,将原始蛋白质序列编码为可由计算机处理、分析的数据。在生物信息学领域,特征的扩展需要通过嵌入进行扩展,从而构建氨基酸词典,如表1所示。每个氨基酸都是一一对应于从小到大的整数,其顺序不会影响实验效果,只是完成字符到整数的转换[16]。由于输入深度学习模型所需的数据规模是固定的,因此必须要进行序列填充。当氨基酸序列“MSFMVPT”的长度为7时,输入长度设置为阈值8,该序列用“X”填充并变为“XMSFMVPT”。
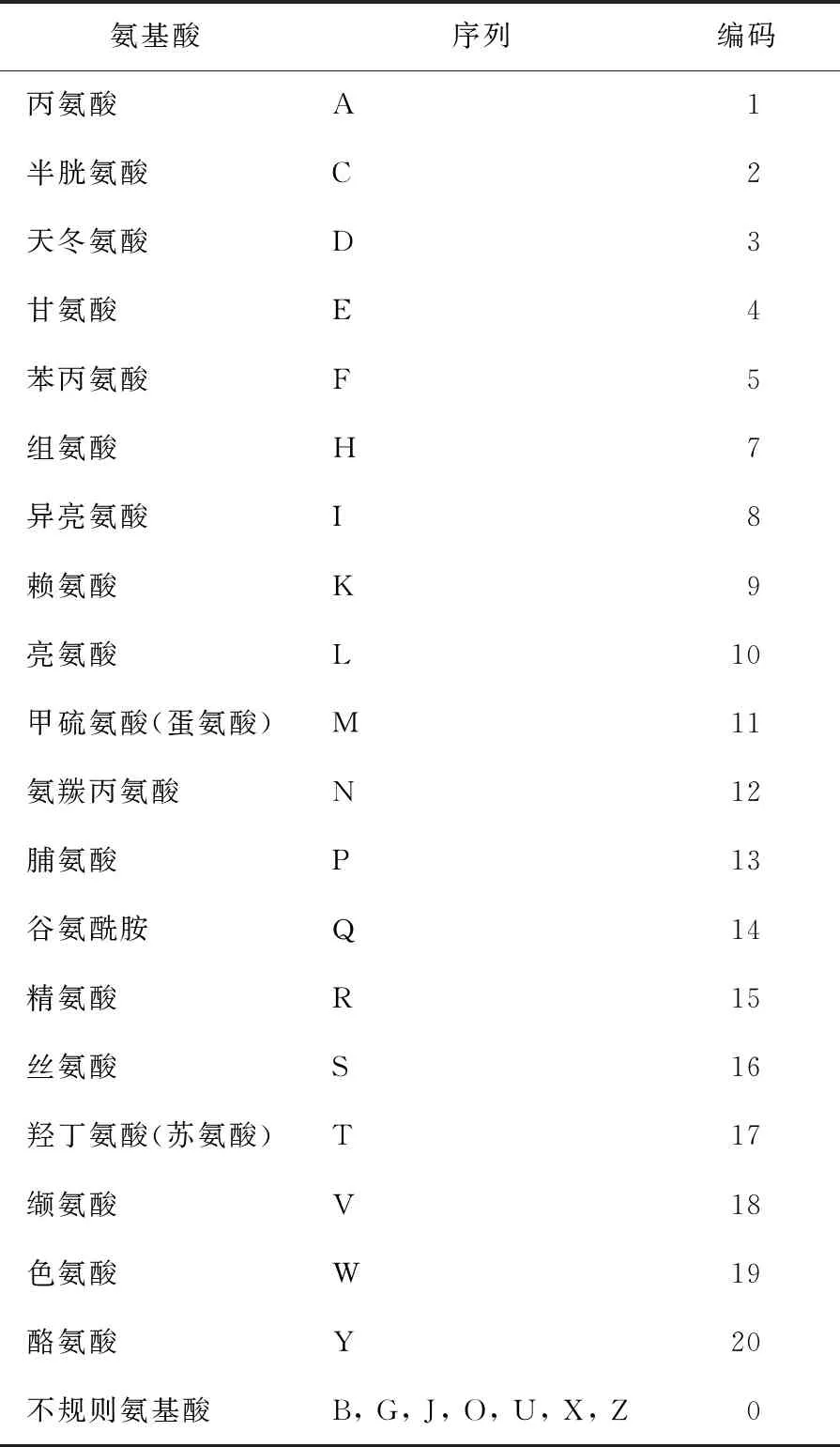
表1 氨基酸词典
2 实验结果与分析
2.1 数据集
使用Zhang等[16]设计的蛋白质资料库PDB14189基准数据集进行实验。该数据集是通过搜索“DNA结合”关键字,并使用UniProt数据库[17]筛选和收集得到的DNA结合蛋白的集合。为了使数据集的品质更加高效,必须筛去全部氨基酸序号小于50且大于6 000的蛋白质序列,还要删除全部不规则氨基酸,如表1中的“X”和“Z”等序列蛋白质。最后,使用BLAST软件对相似程度超40%的序列进行冗余过滤操作[18]。
2.2 评价指标
通过在基准数据集上进行实验,根据实验指标来评估本文算法的优越性,衡量模型的预测效果。由于单一的准确率σacc指标不能完全表征本文算法的预测效果,因此还需要引入其他评估指标,如灵敏度σsen、 特异性σspe等[19-21]。其中,准确率σacc表征算法正确预测的样本的能力,灵敏度σsen表征算法正确预测的阳性样本的能力,特异性σspe表征算法正确预测的阴性样本的能力。评估指标的计算公式分别为
(1)
(2)
(3)
式中:NTP为正确预测的阳性样本的数量;NTN为正确预测的阴性样本的数量;NFP为错误预测的阳性样本的数量;NFN为错误预测的阴性样本的数量。
2.3 对比实验
2.3.1 蛋白质不同尺度特征对比
蛋白质不同尺度特征在PDB14189基准数据上的表现如表2所示。 由表可以看出, 本文算法获得的准确率、 灵敏度、 特异性数值均大于前4个序列的, 表明该算法对基准数据的识别能力更强。为了评估本文算法的预测能力,分别绘制了不同方法(文献[2]、 [22]、 [23]中的方法)的受试者工作特征曲线(ROC)和召回率(PR)曲线,如图2所示。由图可以得出,本文算法在单尺度特征的基础上结合了不同尺度的特征,得到了更有意义的结果。
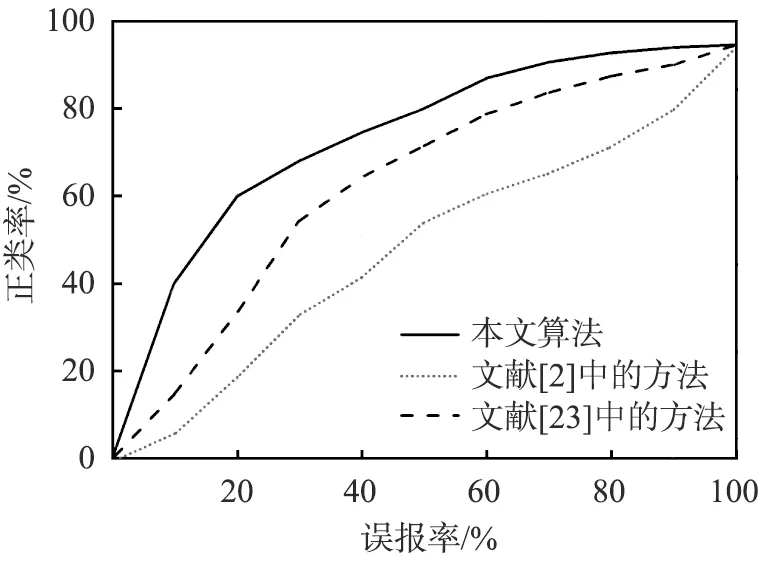
(a) ROC曲线
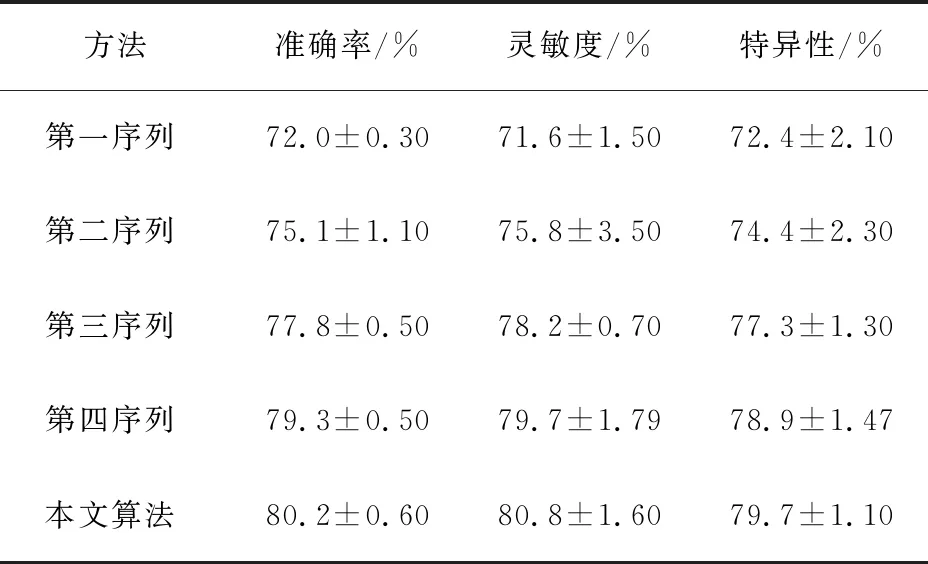
表2 蛋白质不同尺度特征在基准数据上的表现
2.3.2 与传统方法的比较
为了检验本文算法的稳健性,在独立数据集PDB2272上对其进行了评估,结果见表3。由表可以看出,与文献[2]、 [22]、 [23]中的方法相比,本文算法的准确率为66.88%,灵敏度为69.93%, 特异性为65.95%, 3个数值都为最大值, 可见本文算法优于的其他传统方法的, 表现了本文算法的优越性。
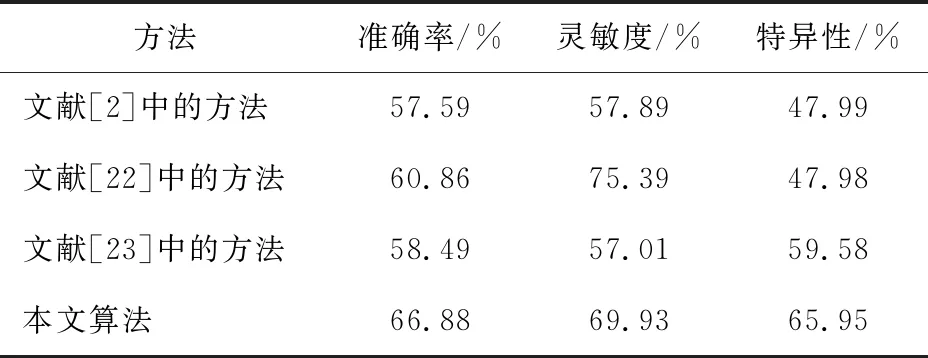
表3 不同算法在3个评估指标上的对比
实际上,非DNA结合蛋白的数量远比DNA结合蛋白的多。本文中基于PDB2272基准数据集进行仿真实验,测试了本文算法的性能,并使用不同的阴性样本与阳性样本的数量比率来进行验证,结果如图3所示。从图可以看出,随着阴性样本数与阳性样本数比率的减小,准确率缓慢增大。在不平衡测试集的情况下,本文算法的性能仍然稳定,并且在DNA结合蛋白的预测中表现良好。
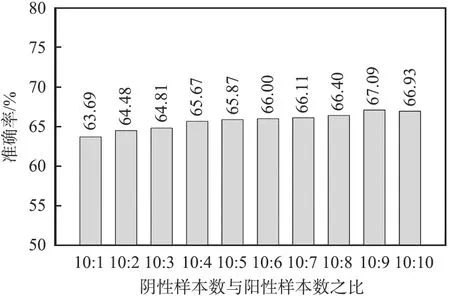
图3 本文算法在PDB2272上的预测准确率
2.3.3 本文算法的应用
为了测试模型的鲁棒性,张戈[7]收集了2 859个蛋白质编号(identity document,ID)。经分析发现,果蝇的2种不同蛋白质的ID对应了相同的蛋白质序列。经过预收集和排序后,获得了2 858个DBP(即DBP2858数据集)。DBP2858数据集中包含人类DBP 的样本数量为1 049,拟南芥(A.thaliana)的为929,小鼠(mouse)的为424,啤酒酵母(S.cerevisiae)的为314,而果蝇(D.melanogaster)的为142。使用PDB14189基准数据集来训练模型,结果如表4所示。在DBP数据集中,本文算法可以正确识别57.83%的蛋白质序列。
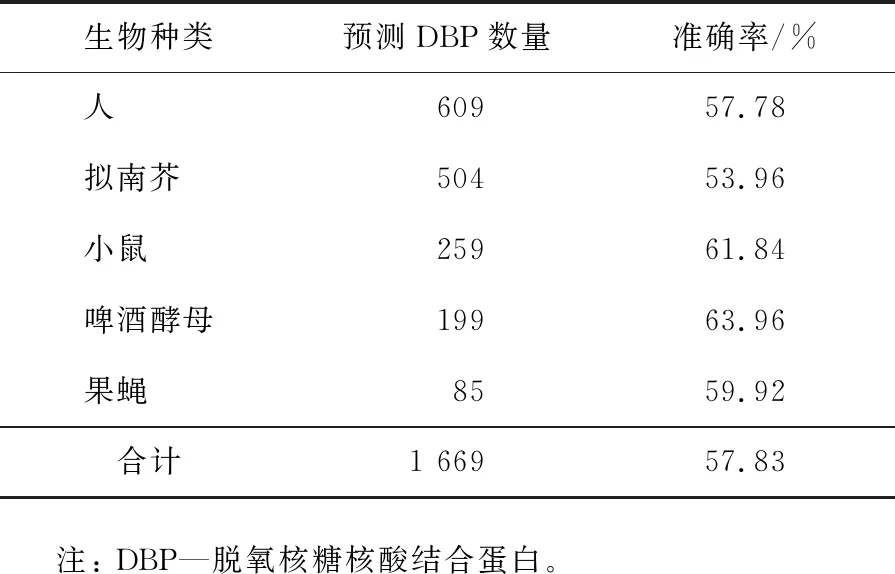
表4 本文算法对不同生物物种的预测性能
3 结论
由于DNA结合蛋白在对生物体的调控机制中具有重要作用, 因此本文中提出了一种基于深度学习和支持向量机的算法用来预测DNA结合蛋白。 在同一数据集上, 分别对本文中提出的深度学习模型和其他传统预测方法进行了训练和实验对比。 实验结果表明, 本文算法对平衡数据集和不平衡数据集都有较好的预测效果, 并且具有较高的预测精度和效率。
