磁性分子印迹电化学传感法检测痕量奥卡西平
2021-08-24李强翔王志梅廖力夫肖锡林
张 迪,李强翔,王志梅,廖力夫,肖锡林,3*
(1.南华大学 化学化工学院,湖南省天然锕系元素配合物设计与应用重点实验室,湖南 衡阳421001;2.宁夏医科大学附属自治区人民医院,宁夏 银川750001;3.湖南大学化学/生物传感与化学计量学国家重点实验室,湖南 长沙410082)
奥卡西平(OXC)是一种可用于局部和全身性癫痫发作治疗的神经性药物,是卡马西平的结构类似物,其副作用比卡马西平小,在临床实践中得到普遍应用。但是,过量使用OXC的患者可能会出现头晕、头痛、皮疹和协调障碍等症状[1-3]。因此,检测体内的OXC血药浓度具有重要价值[4]。现有多种检测OXC的方法,包括分光光度法[5]、表面增强拉曼光谱法(SERS)[6]、液相色谱-质谱/质谱法(LC-MS/MS)[7]、高效液相色谱法(HPLC)、热重分析法[8]、滴定法[9]、薄层色谱法(HPTLC)[10]等,但这些检测方法通常操作复杂,成本较高。电化学传感器因制备简单、价格低廉、灵敏度高[11]等特点而被广泛应用于分析检测。基于此特点,本文创建了一种高灵敏检测样品中OXC的电化学传感器。
不同于分子探针及分子传感器,分子印迹传感器是一种在外层固体基质表面上分布印迹识别位点的器件[12-13]。磁性纳米颗粒(NPs),尤其是Fe3O4@SiO2磁性纳米颗粒(MNPs),由于其顺磁特性和独特的物理化学特性而被应用于许多领域,例如分离、固相萃取、电化学等[14-15]。在应用中,由Fe3O4@SiO2磁性纳米颗粒制备的磁性分子印迹膜可在磁场下快速分离[16]。此外,经二氧化硅包裹的Fe3O4不仅可防止氧化和结块,而且可以大大暴露出电极表面的活性位点,使靶标嵌入的可能性降低,从而为进一步功能化结构修饰提供了可能[14]。
该文中,计算机通过模拟并计算功能单体与不同模板分子的结合能(ΔE),筛选出合适的功能单体组合。在MNPs存在下,由模板分子(OXC)与筛选的功能单体组合(4-VP和MAA)在引发剂的作用下通过自由基交联聚合得到聚合物(OXC/MNPs-MIP),随后将经洗脱处理后的聚合物用于制备电化学传感器MNPs-MIP/MCPE,并用于OXC的检测。
1 实验部分
1.1 试剂与仪器
奥卡西平(OXC)购自本地三甲医院;4-乙烯基吡啶(4-VP)、甲基丙烯酸酯(MAA)、原硅酸四乙酯(TEOS)、2,2′-偶氮双(2-甲基丙腈)(AIBN)、甲酸(MeCOOH)、乙醇(EtOH)、乙二醇二甲基丙烯酸酯(EGDMA,阿拉丁试剂有限公司)。以上试剂均为分析纯,实验用水为二次蒸馏水,所有实验均在室温下进行。
CHI660C电化学工作站(上海晨华仪器有限公司),Agilent 1100高效液相色谱(HPLC,4.6 mm×250 mm,5 μm,安捷伦科技公司),Tecnai G2 F20透射电子显微镜(美国FEI公司)。
1.2 分子印迹聚合物的模拟
基于计算机Gaussian 09程序,在DFT/B3LYP/6-31+G基组条件下,优化出功能单体与OXC的最佳组合形式,计算出功能单体和OXC的ΔE和溶剂化能(ΔE*)。
1.3 MNPs-MIP/MCPE的制备
1.3.1 Fe3O4@SiO2(MNPs)的制备根据参考文献制备Fe3O4[17]:将1.35 g FeCl3·6H2O和3.6 g NaAc在40 mL乙二醇溶液中超声30 min,搅拌直至完全溶解,将该混合物在N2下除空气10 min后转移至聚四氟乙烯高压釜中,于200℃加热8 h得到Fe3O4磁性纳米颗粒,产品分别用乙醇和水洗涤多次,然后在真空干燥箱于60℃下真空干燥12 h。
采用凝胶-溶胶法制备Fe3O4@SiO2[18]:将0.800 g Fe3O4分散在无水乙醇-水(体积比为4∶1)溶液中,超声30 min。然后将5 mL氨水和5 mL TEOS逐滴加入上述溶液中,室温下搅拌6 h,反应结束后在磁铁的作用下用水和无水乙醇洗涤数次后分离出产物,置于60℃真空干燥8 h后,得到具有核壳结构的Fe3O4@SiO2磁性纳米粒子。
1.3.2 磁性分子聚合物(MNPs-MIP)的制备根据参考文献[19],在含有40 mL乙醇的烧杯中溶解1 mmol OXC和2 mmol MAA,搅拌反应2 h,再加入1 mmol 4-VP,搅拌反应1 h。然后加入0.05 g Fe3O4@SiO2、4 mmol EGDMA(交联剂)和60 mg AIBN(引发剂),氮气置换除氧密封,超声60 min,40℃下反应12 h得到OXC/MNPs-MIP。最后,将制备的物质置于乙酸-甲醇(体积比为3∶7)洗脱液中洗脱60 min,获得MNPs-MIP并用水洗涤数次,在60℃真空干燥8 h,即得MNPs-MIP。
1.3.3 磁性碳糊电极的组装称取4 g石墨粉,1 mL石蜡油在烧杯中混合并搅拌至糊状物,取适量糊状物紧压入长度约为8 cm内径为4 mm的聚丙烯塑料管中,管的末端插长约5 cm的铅笔芯作为电路连接点,另一端约1.5 mm处嵌入一块圆形铷铁硼磁铁(直径4 mm,厚为2 mm),再填充石墨粉包覆后打磨光滑,即制得磁性碳糊电极(MCPE)[20]。
将1.0 mg MNPs-MIP超声分散在1 mL乙醇中,取其分散均匀的25 μL悬浮液均匀滴加到MCPE表面,得到MNPs-MIP/MCPE传感器(如图1A)。图1B表示分子印迹聚合物的洗脱-重新结合机制,其中,分子印迹聚合物的洗脱和重结合是模板分子与功能单体混合后在氢键作用下形成的。作为对比,本文通过类似的操作制备了裸电极(MCPE),表面仅装有MIP的磁性石墨电极(MIP/MCPE)及不加OXC制备的非分子磁性印迹聚合物MNPs-NMIP/MCPE。

图1 MNPs-MIP/MCPE传感器的设计和识别示意图(A),及分子印迹聚合物的洗脱-重新结合机制(B)Fig.1 The schematic diagram for design and identification of the MNPs-MIP/MCPE sensors(A)and elution-rebinding mechanism of molecularly imprinted polymers(B)
1.4 电化学检测
经修饰的MCPE与铂电极和甘汞电极组成三电极系统,通过电化学工作站检测OXC。将工作电极MCPE在OXC标准溶液中重新混合15 min,电极置于5 mL PBS缓冲液(pH 7.2)中进行检测。实验在DPV的最佳参数下进行:电势从-1.5 V到-0.8 V,脉冲幅度为50 mV,扫描速率为30 mV/s,脉冲宽度为170 ms,电势增量为20 mV。
1.5 实际样品的制备
奥卡西平片(300 mg/片)、奥卡西平口服混悬液(60 mg/mL)在当地三甲医院获得。将一片用研钵粉碎的片剂溶于30 mL乙醇或5 mL口服混悬液加至25 mL乙醇。通过0.22 μm过滤器过滤后,将滤液转移至50 mL容量瓶中,并用乙醇稀释至刻度。然后,将100 μL该溶液加至5.0 mL PBS(pH 7.2)中,用作测试溶液。
收集志愿者服用300 mg OXC 3 h后的尿液样本。在每次检测前,将2 mL尿液与2.0 mL甲醇混合,10 000 r/min离心15 min以去除蛋白质。然后将上清液用PBS(pH 7.2)稀释至5.0 mL,以降低基底效应,随后用于分析。
2 结果与讨论
2.1 分子优化
由于功能单体和溶剂的选择影响MIP印迹位点的形成和识别性能[21-23],且多种功能单体制备的MIP比单一单体具有更好的选择性[24]。根据密度泛函数理论(DFT/B3LYP/6-31+G法),实验优化了模板分子OXC和不同功能单体的最优构型(图2),随后依据图3所示分别将不同的功能单体与模板分子结合计算其结合能ΔE,计算公式为:ΔE=ECET-ΣEM,式中,ΔE为聚合物的结合能,EC为聚合物的总能量,ET为OXC的能量,ΣEM为游离功能单体的能量之和。ΔE越小表示其结合能力越强,从表1(1~6)中不同组合构型的ΔE中得出以4-VP和MAA作为功能单体时,制备的印迹聚合物效果最佳。因此,实验选择OXC-(4-VP)-MAA为最佳体系。究其原因,可能是因为4-VP中的N可与OXC的羰基氧形成氢键,同时,4-VP的吡啶环与OXC的苯环可形成“π-π堆积”,从而大大提高了MIP结构的稳定性。而MAA不仅是质子供体,还是质子受体,可通过与OXC形成氢键和离子键以增强MIP结构的稳定性[19,25]。

表1 模板分子(OXC)与各不同功能单体复合物的结合能(ΔE)Table 1 The binding energy(ΔE)of different template molecule(OXC)-functional monomer complexes

图2 功能单体及模板分子OXC的结构优化图Fig.2 Structure optimization diagrams of functional monomers and the template molecule OXC

图3 模板分子OXC结合单功能单体的结构优化图Fig.3 Structure optimization diagrams of the template molecule OXC combined with single functional monomer
随后,根据OXC分子的静电势图(见图4)可得出处于两个不同位置的羰基氧的电负性不同(a=-0.461,b=-0.530)。因此,探讨了OXC与功能单体的结合位点和组分比(图5,表1中7~10)。在聚合物制备过程中,首先在体系中添加MAA,其羧基氢与OXC的羰基形成氢键—O…H—O—CO,使OXC结构中a、b位置的羰基氧被占据,而后体系中加入4-VP,4-VP的氮与OXC的氨基结合形成—CO—NH2…N—。
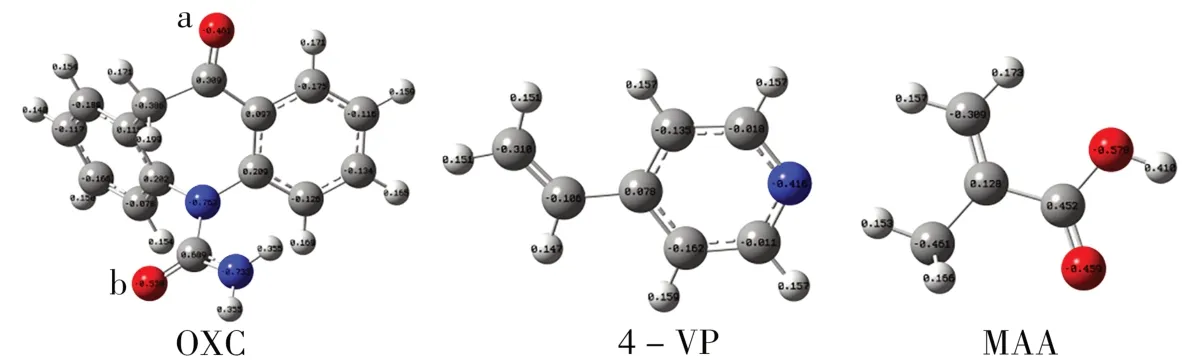
图4 模板分子OXC、功能单体(4-VP和MAA)的静电势图Fig.4 The electrostatic diagrams of the template molecule OXC and functional monomers(4-VP and MAA)

图5 功能单体(4-VP和MAA)以不同的组分比结合模板分子(OXC)的结构图Fig.5 Structure diagrams of the template molecule OXC and functional monomers(4-VP and MAA)combined in different component ratios
此外,如表1(10)所示,在OXC+(4-VP)+MAA=1∶1∶2时,ΔE最小,表明此时聚合物相对最稳定。因此推断模板分子OXC与功能单体4-VP和MAA的结合比可能为1∶1∶2。
此外,反应溶剂也是反应成功的关键因素,溶剂化能|ΔE*|越小,对功能单体与模板分子的结合越有利。|ΔE*|计算公式为:|ΔE*|=Es-Ev,式中,|ΔE*|为聚合物的溶剂化能,Es为聚合物在溶剂环境中的结合能,Ev为聚合物在真空环境中的结合能。选择的溶剂不仅需对印迹分子具有好的溶解性,且不能参与OXC与功能单体之间的结合。在制备聚合物的过程中,溶剂与聚合物之间的作用力越弱,越有利于聚合物的制备。本文模拟了聚合物分别以苯、丙酮、乙醇、二甲基亚砜(DMSO)和水作为溶剂的结合效果,测得聚合物的|ΔE*|分别为15.411、46.225、47.144、49.227、50.200,即|ΔE苯*|<|ΔE丙酮*|<|ΔE乙醇*|<|ΔEDMSO*|<|ΔEwater*|。但由于苯不溶解OXC,而丙酮毒性相对较大。因此,实验最终选择价格低廉,且能快速溶解OXC的乙醇作为溶剂。
2.2 Fe3O4纳米颗粒磁性印迹聚合物的表征
分别考察了Fe3O4、MNPs和MNPs-MIP的TEM图像(图6A~C)。通过对3图的比较,可看出Fe3O4被SiO2成功包裹,形成了Fe3O4-SiO2核-壳结构,且Fe3O4-SiO2核壳纳米颗粒的尺寸较大,这可能是在包硅过程中Fe3O4纳米颗粒出现团聚,从而出现了“多核”的Fe3O4-SiO2核壳纳米颗粒。

图6 Fe3O4(A)、MNPs(B)及MNPs-MIP(C)的TEM图Fig.6 TEM images of Fe3O4(A),MNPs(B)and MNPs-MIP(C)
2.3 MNPs-MIP/MCPE传感器的电化学表征
在80 μmol/L OXC溶液中,考察了MNPs-MIP/MCPE和MCPE的循环伏安(CV)图(图7)。结果显示,对于MCPE,OXC的峰值电流很弱(曲线a)。对于MNPs-MIP/MCPE电极,可观察到在-1.19 V处出现1个峰电流值更大的阴极峰(曲线b)。表明已制备出MNPs-MIP/MCPE传感器,且MIP与OXC具有良好的配位作用。

图7 MCPE(a)及MNPs-MIP/MCPE(b)在OXC溶液中的循环伏安图Fig.7 CV curves of MCPE(a)and MNPs-MIP/MCPE in OXC solution
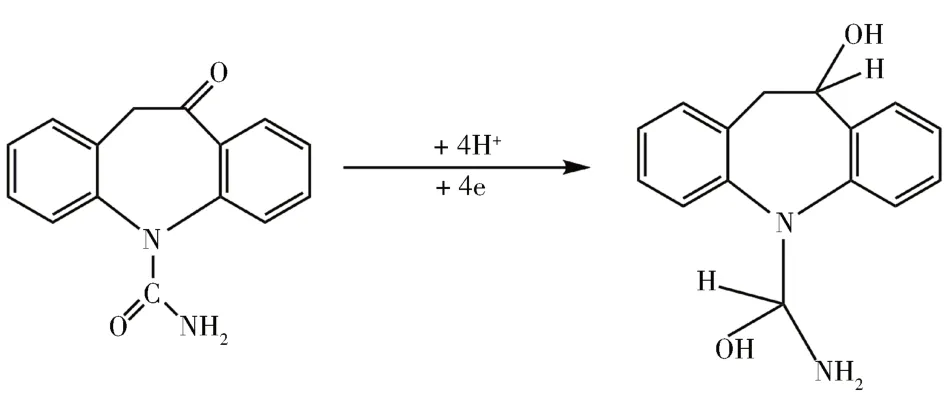
对于MNPs-MIP/MCPE电极在-1.19 V处的阴极峰,根据OXC的结构推断,发生的反应机理可能如下:
用DPV法进一步探究经MNPs-MIP修饰的电极及不同干扰电极上OXC的峰电流信号。如图8(a-e)所示,OXC/MNPs-MIP/MCPE上的-1.19 V处出现明显的峰值电流(图8a)。在OXC/MNPs-MIP/MCPE上洗脱后,电流值较低,表明OXC已基本洗脱(图8d)。在80 μmol/L OXC溶液中重组后,研究MCPE(f)、MNPs-MIP/MCPE(b)、MIP/MCPE(c)和MNPs/MCPE(e)的电流信号强度。结果显示,在MNPs-MIP/MCPE上显示出较高的电流值(图8b)。上述实验进一步证明印迹孔对目标具有较高的识别能力,且相对于MCPE(图8f)和MNPs/MCPE(图8e),OXC在MNPs-MIP/MCPE的电势值有正向偏移,可避免某些干扰物质的影响。
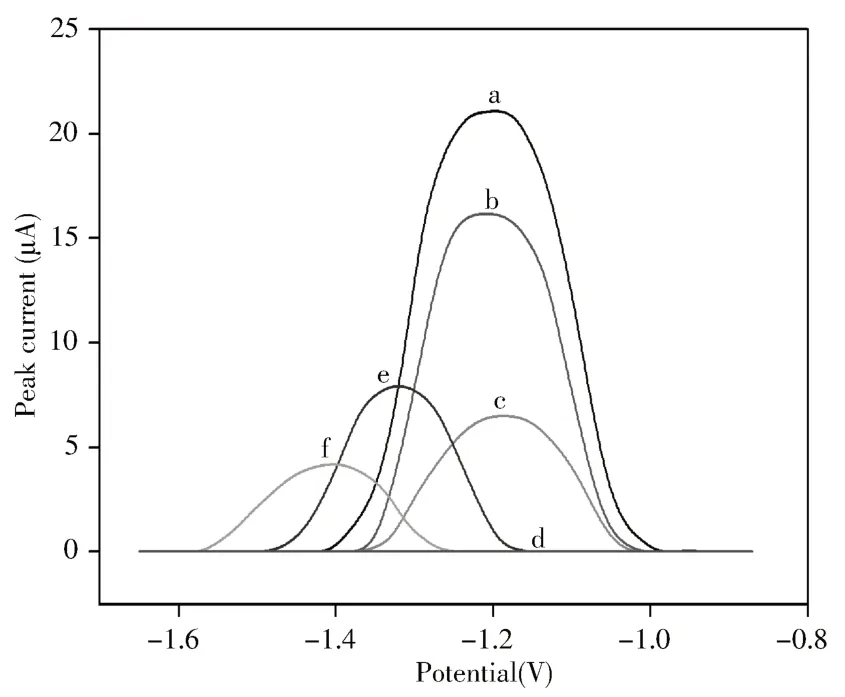
图8 不同电极在PBS(pH 7.2)溶液中的DPV曲线Fig.8 DPV curves of different electrodes in PBS(pH 7.2)solution
采用电化学阻抗法进一步研究不同修饰电极在含有2.0 mmol/L K3Fe(CN)6/K4Fe(CN)6的0.2 mol/L KCl中的电化学性能(图9)。结果显示,裸电极MCPE的阻抗最小(曲线a),曲线几乎呈一直线,表明电极表面可能为扩散限制过程[26-27]。MNPs-MIP/MCPE的阻值相对较小(曲线b)。将MNPs-MIP/MCPE浸 入80 μmol/L OXC溶液中15 min,Rct值增至824 Ω(曲线c),表明探针分子通过的某些通道被阻断。OXC/MNPs-MIP/MCPE的阻抗为1487 Ω(曲线d),表明已成功构建了聚合物层。洗脱靶后,电荷转移电阻(Rct)降至157 Ω,这是因为洗脱后分子在电极上留下了空穴,从而减小了阻值。MNPs-NMIP/MCPE(曲线e)的Rct值为2 764 Ω,表明由于电极表面无印迹孔,探针分子几乎无法通过。

图9 在含有2.0 mmol/L K3Fe(CN)6/K4Fe(CN)6的0.2 mol/L KCl中不同电极的EIS图Fig.9 EIS curves of different electrodes in 0.2 mol/L KCl containing 2.0 mmol/L K3Fe(CN)6/K4Fe(CN)6
2.4 实验条件的优化
2.4.1 洗脱溶剂与洗脱时间的选择洗脱液的选择是实验的关键因素。洗脱液的洗脱强度太大,会洗脱整个印迹;洗脱力度太小,则印迹空腔洗脱不完全。本文基于经修饰的MCPE与铂电极和甘汞电极组成三电极系统的DPV法在PBS缓冲液中考察了洗脱剂种类与配比的影响。考察了甲醇-乙酸(9∶1、8∶2、7∶3)、甲醇、乙醇和十二烷基硫酸钠(SDS)分别用作洗脱液时的响应信号。结果显示,以甲醇-乙酸(7∶3)为洗脱溶剂时,实验效果最佳。
由于重组时间过长易造成不必要的时间损失,时间过短则实验效果将受到较大影响,本实验在50.0 μmol/L OXC溶液中考察了MNPs-MIP/MCPE和MNPs/MCPE的重组时间。如图10所 示,与MNPs/MCPE电 极 相 比,OXC在MNPs-MIP/MCPE表面上显示出较高的电信号,表明印迹聚合物对模板分子有较为显著的识别能力,且Fe3O4@SiO2与分子印迹膜相互作用使该传感器具有更高的选择性。此外,MNPs-MIP/MCPE的峰值电流随着时间的延长而增加,且该电流在15 min内几乎饱和。为了保证实验的重组反应时间和高效性,选择15 min作为最佳重组时间。
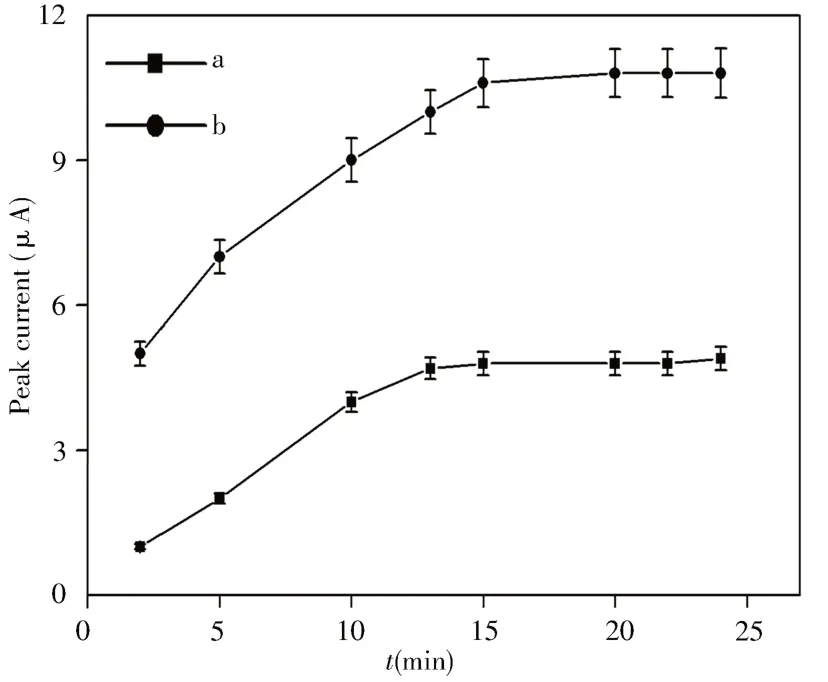
图10 MNPs/MCPE(a)和MNPs-MIP/MCPE(b)对OXC响应时间的优化Fig.10 Time optimization on the response of MNPs/MCPE(a)and MNPs-MIP/MCPE(b)
2.4.2 缓冲溶液pH值的选择电解质的pH值也会对电极表面的峰电流(IP)与峰电压(Ep)有较大的影响,进而影响传感器的灵敏度。本实验将MNPs-MIP/MCPE置于100 μmol/L OXC标准溶液中重结合后,选择pH=4.0~8.5的PBS缓冲液进行考察(图11)。图中可以清楚地观察到Ep随着pH值的增加而线性递减,线性方程为:Ep=-0.748 0-0.061 7 pH(r=0.999 6),斜率值近似为能斯特方程计算的理论值-0.059V/pH。说明OXC参与反应的质子数和电子数相等,且pH 7.2时峰电流最大。因此,本文选择在pH 7.2的PBS缓冲液中检测样品中OXC的浓度。

图11 PBS缓冲液的pH值对OXC的电势(Ep)和峰电流(Ip)的影响Fig.11 Effect of pH value on potential(Ep)and peak current(Ip)of OXC in PBS buffer
2.5 MNPs-MIP/MCPE的性能研究
2.5.1 线性范围与检出限在上述最优条件下,采用MNPs-MIP/MCPE传感器对不同浓度(5×10-8~1.5×10-4mol/L)OXC的DPV信号进行检测。如图12所示,传感器的峰电流信号随OXC浓度的增大而增大,且OXC分别在5×10-8~3×10-6mol/L和3×10-6~1.5×10-4mol/L浓度范围内与其峰电流信号呈线性关系,其线性方程分别为:Ip(μA)=1.755+1.097c(μmol/L),相关系数(r)=0.999 7和Ip(μA)=0.131+5.177c(μmol/L),r=0.999 6。中间线性的中断可能是由于OXC在第一线性范围内亚单分子的形成以及在第二线性范围内单分子的形成所致[28-29]。传感器检测OXC的 检 出 限(LOD)为2.06×10-8mol/L(LOD=3S/m,其中S为校准曲线截距的标准偏差,m为校准曲线的斜率)。
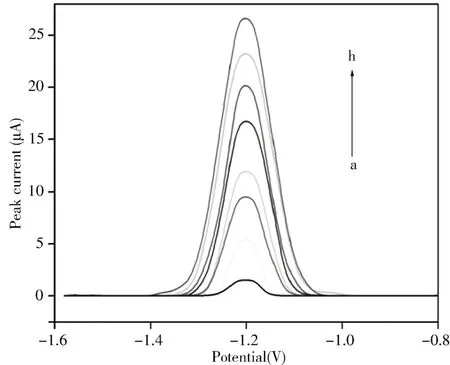
图12 MNPs-MIP/MCPE传感器在不同浓度OXC中的DPV曲线Fig.12 DPV curves of MNPs-MIP/MCPE in different concentration of OXC concentration of OXC(a-h):1.0,2.5,30.0,50.0,90.0,110.0,130.0,150.0 μmol/L
2.5.2 稳定性与选择性分别在20、40、60 μmol/L OXC溶液中对同一MNPs-MIP/MCPE传感器的峰电流进行5次分析,3种OXC浓度的RSD分别为4.8%、5.3%、5.0%。同一传感器在存放30 d后,其电化学响应信号仍为初始值的95.3%。实验结果表明该传感器具有较好的重现性和稳定性。
此外,研究了传感器对OXC的选择性和其他干扰物质对检测OXC的影响。结果表明,当OXC的浓度为1 μmol/L时,5倍浓度的类似结构药物卡马西平(CBZ)和拉莫三嗪(LMT);200倍浓度的10倍浓度的尿酸(UA)、多巴胺(DA)、维生素(VC);15倍浓度的苏氨酸(Thr)、亮氨酸(Leu)、天冬氨酸(Asp),对OXC的检测影响均在允许相对误差(10%)以内。表明本文制备的MNPs-MIP/MCPE传感器对OXC的检测具有较高的选择性。
2.6 实际样品的分析
基于本方法检测了片剂(300 mg/片)、口服混悬液(60 mg/mL)及尿液中的OXC含量[30],以评估MNPs-MIP/MCPE传感器的检测性能,结果如表2所示。通过加标回收实验考察了该方法的准确度,即向样品中分别加入20、40、60 μmol/L的OXC,每一浓度样品平行检测3次,测得回收率为99.4%~101%,相对标准偏差(RSD)为1.5%~2.5%。实验结果表明,该方法具有良好的精密度和准确度。进一步采用HPLC法进行验证,实验结果在置信度95%以上,表明该传感器可用于实际样品中OXC的检测。

表2 MNPs-MIP/MCPE传感器对片剂、口服混悬液和尿液中的OXC检测结果(n=6)Table 2 Detection result of OXC in tablets,oral suspension and urine by MNPs-MIP/MCPE sensor(n=6)
将该方法与其他电化学检测方法进行比较(如表3),可知本文建立的传感器具有更低的检出限和更宽的线性范围,是一种成本更低、灵敏度更高的检测工具。

表3 该方法与其他电化学方法测定OXC的对比Table 3 Comparison of this method with the other electrochemical methods for the determination of OXC
3 结论
本文合成了一种磁性印迹传感器MNPs-MIP/MCPE用于检测样品中的OXC。实验首先通过模拟计算筛选出最佳功能单体组合(4-VP和MAA)及反应溶剂,并基于磁性Fe3O4@SiO2NPs为原料,合成了磁性印迹传感器MNPs-MIP/MCPE。结果显示,体系酸度为7.2的条件下,MNPs-MIP/MCPE传感器具有出色的检测OXC的性能,可实现对实际样品中OXC的测定。该传感器制备简单、灵敏度高,可为其他检测方法提供一定的理论基础。
