Bi2Te3基热电材料的电输运性能研究进展
2021-08-19王晴刘子杨赵沙沙李志亮
王晴,刘子杨,赵沙沙,李志亮
(河北大学 物理科学与技术学院,河北省量子场论高精度计算与应用重点实验室,河北 保定 071002)

自20世纪20年代以来,伴随着半导体物理与固体物理等基础理论的发展,热电材料相关研究再次引起全球关注,人们发现半导体材料不仅具有较高的塞贝克系数,其导热机制也与金属不同[6].近半个世纪以来,热电材料的发展取得快速进步,一些具有优良性能的热电材料相继被开发出来,如方钴矿、黄铜矿、GeTe合金、PbTe合金等.zT值也从早期约为1.0提高到了2.0及以上.尽管如此,Te基材料,特别是Bi2Te3基材料,仍然是目前商业化应用最广泛的材料之一.该类材料之所以备受商业化应用的青睐,主要源于其独特的能带结构所赋予的优良热电性能.1965年,Greenaway等[7]发现,Bi2(Te,Se)3材料中Se的含量增大时,带隙逐渐增大,当化学计量比达到Bi2Te2Se时,带隙出现极大值,但当Se含量继续增加,带隙则减小.另外,当载流子浓度较大时,等能面的形状也会随载流子浓度的变化而变化,载流子的态密度有效质量也会因此受到影响,进而影响其热电性能.其次,Bi2Te3基材料中存在大量点缺陷,如反位缺陷和空位,而晶体内部的缺陷会对载流子及声子的输运造成影响,从而影响热电性能.1957年,Satterthwaite等[8]发现,Bi2Te3中元素含量的改变可以实现材料N型和P型转换,当Te的摩尔分数高于62.8%时为N型,而且随着Te含量的增多,材料的电子浓度提高;反之,当Bi过量时,材料为P型,这种现象与Bi2Te3晶体内部的点缺陷有关.因此,可以通过将Bi2Te3与同结构的Sb2Te3或Bi2Se3合金化以增加声子散射,并优化能带结构,从而提高热电性能[9].
能带结构对于优化(Bi1-xSbx)2(Te1-ySey)3的热电性能起着重要作用,因此如何准确计算能带结构从而调控(Bi1-xSbx)2(Te1-ySey)3材料的热电性能非常重要,(Bi1-xSbx)2(Te1-ySey)3中Se、Sb含量的改变是引起的能带结构变化的主要因素,值得归纳和探讨.该综述总结了近年来关于Bi2Te3基热电材料的研究进展,讨论了影响其能带结构的关键因素,归纳出了能带结构与电输运性能间的关系和部分调控规律.
1 Bi2Te3的晶体结构简介


图1 Bi2Te3的晶体结构示意Fig.1 Crystal structure of Bi2Te3
2 Bi2Te3的电子结构和电输运性能
Bi2Te3是目前近室温区应用最广泛的热电材料,关于Bi2Te3电子结构的相关研究颇多.根据Bi2Te3能带结构的早期实验研究,通过Shubnikov-de Haas和de Haas-van Alphen测量,六能谷模型已经被人们普遍接受[12-14],测量得到Bi2Te3的价带顶(VBM)和导带底(CBM)的简并度Nv=6.为了解释六能谷模型,人们对Bi2Te3的能带结构进行了许多计算研究.
2.1 利用实验晶格常数计算能带结构
人们早期利用经验赝势法来计算Bi2Te3的能带结构.1992年,Thomas等[15]在基于线性缀加平面波(LAPW)的基础上利用密度泛函理论(DFT)计算了Bi2Te3的能带结构,结合自旋轨道相互作用,解释了Bi2Te3带隙形成的物理原理[16-17].之后,Mishra等[18-19]利用局域密度泛函(LDA)并考虑了自旋轨道相互作用的影响,计算并获得了Bi2Te3能带结构,求出来VBM的简并度Nv=6,与Shubnikov-de Haas和de Haas-van Alphen的测量结果一致[12,20].但CBM的简并度Nv=2,与实验结果不符[13-14],这可能是由于计算忽略了非球面势能的影响.
然而,2000年,Larson等[21-23]使用LAPW和广义梯度近似法(GGA-PBE)计算出CBM有2个能谷.他们认为之所以未在CBM中找到六能谷,是由于自旋轨道相互作用(SOI)或对有限温度效应处理不准确所导致,这2种情况有可能改变CBM的位置.
2001年,Youn等[24]也利用LAPW计算了Bi2Te3的能带结构,但他们得到的VBM和CBM均有6个能谷.不同的是,Youn等使用LDA替换了PBE,这可能导致计算中交换关联泛函的不同,进而使计算结果也不同.此外,他们将Te的4p电子作为核心态,而Larson等[25-26]将Te的4p电子作为价态处理.Larson等的理论计算结果表明,使用Youn等的方法不能得到CBM的简并度Nv=6.此外,他们采用了p1/2校正以及线性缀加平面波结合局域轨道方法(LAPW+LO)和GGA,最终求得VBM和CBM的简并度Nv=6.他们提出,p1/2校正在修正CBM简并中起到重要作用.但是,Scheidemantel等[27]指出,仅使用LAPW+LO和GGA来计算Bi2Te3能带结构,求得VBM和CBM的简并度Nv=6,所以p1/2校正并不是必需的.他们认为能谷的数量取决于LAPW的第二变分公式中的能量窗口,当能量窗口低于6 Ry时,CBM的简并度Nv=2,当能量窗口大于6 Ry时,CBM的简并度Nv=6.
根据实验晶格参数,使用不同计算方法得到的能带结构在带隙和能带边缘上有很大差异.如表1所示,这些计算结果可分为3类,第1类结果具有合适的带隙值,接近于0.13~0.17 eV的实验值[20,28-29],但这是以牺牲CBM的简并性为代价的.第2类计算结果虽然与实验提出的六能谷模型一致,但却低估了带隙值.第3类计算结果与实验值相符,这可能是由于选取了合适的能量窗口值.
2.2 范德华力对能带结构产生的影响
2002年,Wang等[30-31]使用带有GGA-PBE的LAPW计算了弛豫的Bi2Te3的能带结构.而弛豫的Bi2Te3的晶格参数和能带结构与未弛豫的Bi2Te3很相似,弛豫和未弛豫的Bi2Te3都存在六能谷模型.Luo等[32]报告了Bi2Te3的晶格参数,尤其是层间距d,即相邻QL中Te(1)原子之间的垂直距离,会随着其他参数的改变而有很大差异.弛豫的Bi2Te3在LDA计算下的d值与实验结果一致,但是GGA-PBE高估了Bi2Te3的d值,因此计算得到的Bi2Te3的弹性常数结果与实验结果有差异[33].
由以上情况可见,晶格参数的改变对Bi2Te3的能带结构影响很大.考虑到晶格弛豫,与GGA-PBE相比,使用LDA与实验测得的能带结构更相符.这是因为使用GGA-PBE时人为压缩了Bi2Te3中范德华键合间隙[33].使用GGA弛豫晶格参数时选择合适的范德华泛函可以修正该类计算误差.理论上,在借助实验晶格参数计算出的不同泛函的能带结构应相同.然而,实际计算所得的能带结构在不同情况下仍有较大差异,如表1所示.

表1 Bi2Te3能带结构的计算方法、带隙、VBM和CBM的简并度Tab.1 Calculated method,bandgap,and degeneracy(Nv)of VBM and CBM for Bi2Te3
2.3 应变与压力对能带结构产生的影响
2016年,管建祥等[34]分别计算了Bi2Te3块体和Bi2Te3单QL薄膜在应变下的电子结构变化,计算结果表明应变并未改变Bi2Te3块体的基本特性,但随着应变的增加,带隙宽度从0.210 eV增加到0.299 eV.并且在应变作用下,导带底的能量水平变化不大,但价带顶的能量变化幅度较大.对于Bi2Te3单QL薄膜,应变并未改变单QL薄膜的间接带隙特性,不过随着应变的增加,价带部分能量区间随之减小,带隙减小.这是由于应变影响了薄膜的结构.另外,随着应变的增加,虽然导带底和价带顶的位置并未改变,但是相对于价带,导带底能量水平变化较大,与Bi2Te3块体情况正好相反,这也是由于应变带来的势场调整对整个电子结构的影响.总之,应变会导致材料的周期势场改变,以致影响材料的能带结构.
2017年,赵堃等[35]的计算结果表明,随着压力的增大,价带顶和导带底的电子能态密度呈现减小的趋势,Bi2Te3的能带展宽变大,这意味着原子间相互作用随着压力的提高而增强.且Bi2Te3的价带随着压力的提高会发生电子的拓扑转变,进而导致其输运特性的反常.
2.4 考虑激发态电子的能带结构计算
近年来,Bi2Te3由于具有拓扑绝缘体(TI)的特性而得到了广泛的研究[36-41].激发态性质,例如准粒子能量和带隙,是拓扑绝缘体研究中的关键性质.DFT计算可以充分描述基态特性,但不适用于描述激发态特性.在计算Bi2Te3的能带结构时对激发态的特性进行不断优化会使结果更贴近实验.
2005年,Kim等[42-44]使用屏蔽交换局域密度近似(sX-LDA)的方法计算了未弛豫的Bi2Te3的能带结构,该法可以较好地描述其他半导体和绝缘材料的激发态特性.Kim等的计算结果表明,VBM和CBM的简并度Nv=6,与六能谷模型一致,所得带隙为0.154 eV,也与实验相符.
准粒子(GW)近似校正法可以精确地描述激发态的性质.GW校正法依据不同的自洽计算方式分为 G0W0(一次自洽计算叠代)、GW0(部分自洽计算叠代)、GW(全自洽计算叠代).对于大部分半导体材料带隙的计算,G0W0会得到比实验值略小的结果,GW0会得到与实验值相接近的结果,完全自洽收敛的GW得到的带隙值则会比实验值略大[45-48].在实际应用中,GW准粒子能量通常以微扰的方式从某个参考单粒子的哈密顿量为出发点计算得到,这就是所谓的G0W0方法.Kioupakis等[49]用LDA+G0W0计算了未弛豫的Bi2Te3的能带结构,得到未经GW校正的带隙为0.087 eV,经GW校正后的带隙增加到了0.165 eV,与实验相符.2013年,Nechaev等[50-51,14,24]用GGA+G0W0和LDA+G0W0来计算弛豫与未弛豫的Bi2Te3的能带结构.他们用GGA+G0W0计算的弛豫的Bi2Te3的能带结构与角分辨光电子发射能谱法(ARPES)所得的实验结果相符,但是VBM和CBM的有效质量张量与Shubnikov-de Haas的测量值略有不同.
尽管使用GW校正计算的带隙接近实验值,但计算非常耗时.修正贝克-约翰逊势(mBJ)不仅在计算精度上不逊于GW校正,而且mBJ耗时更短、成本更低[52].2015年,Shi等[53]使用LDA+mBJ计算了Bi2Te3的能带结构,得出带隙为0.14 eV,与实验结果[28-29,31]和GW计算结果一致[49-50].更重要的是,他们提出了热电性能与拓扑绝缘体中电子行为之间的关联性.SOI作用引起的能带反转是拓扑绝缘行为所必需的,复杂的能带结构(例如高度各向异性的载流子有效质量和多重简并的能带结构)有利于提高热电性能,也与能带反转有关.Heremans等[54]使用同样的方法来计算弛豫的Bi2Te3的能带结构和费米能级.计算得到弛豫Bi2Te3的带隙为0.15 eV,接近Shi等[53]的计算结果.根据能带结构,VBM和CBM分别位于ZF和ZΓ方向.载流子浓度为1.1×1019cm-3时,N型和P型Bi2Te3的费米能级的计算结果与ARPES的数据吻合较好[51].
2.5 Bi2Te3的电输运性能
电输运性能包括塞贝克系数与电导率2方面,当材料是简并半导体或重掺杂时,采用玻尔兹曼方程结合弛豫时间近似模型,电输运性能的表达式

式中下标n、p分别是电子和空穴的作用成分,Sn是负值,Sp是正值.上述公式表明塞贝克系数和电导率是载流子浓度和有效质量、散射系数、费米能级等诸多参数构成的函数.它既体现了与电输运性能相关的各种因素,又阐明了热电参数之间的矛盾-耦合关系.对于Bi2Te3材料而言,存在一个最优载流子浓度,使其zT值达到最大值.2016年,Yim等[55-56]提出该体系的最优载流子浓度约为2×1019cm-3,此时塞贝克系数大概为200 μV/K,电导率大概为105S/m,后续的研究以及理论计算也证明了这一点[55,57].
进一步深入的理论计算研究表明,利用对Bi2Te3材料能带结构的改变,可以提高材料的能带简并度.能带简并度增大,能够增加参与电传输的能谷数,材料的态密度有效质量也会增大[58],从而提高塞贝克系数[59-60].Jaworski等[61]通过向P型Bi2Te3中掺入Sn元素引入共振能级,使材料拥有更大的塞贝克系数,从而改善了材料的电输运性能.
3 Bi2Te3-xSex的电子结构和电输运性能
纯的Bi2Se3材料为N型半导体,关于Bi2Te3-xSex材料,当x达到一定值时,材料会由N型变为P型[62].Se代替Te对Bi2Te3的带隙类型和能带结构均会产生影响,相关研究值得进一步讨论.
3.1 Bi2Se3二元化合物简介

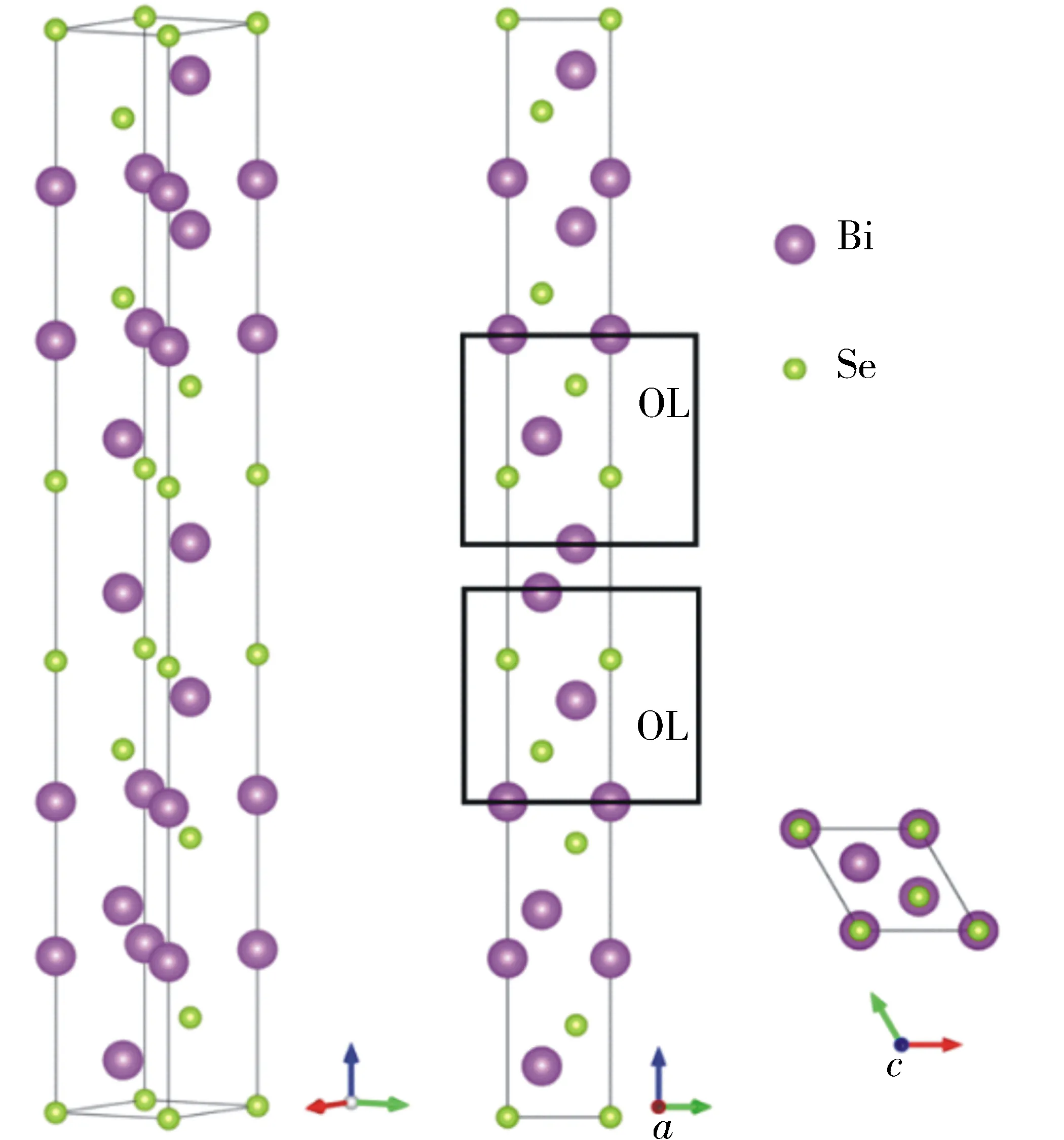
图2 Bi2Se3的晶体结构示意Fig.2 Crystal structureof Bi2Se3
对于Bi2Se3带隙结构的主要争论点在于是直接带隙还是间接带隙.在不考虑自旋轨道耦合的情况下,Bi2Se3是直接带隙.但若考虑强自旋轨道耦合,Bi2Se3则为间接带隙.
1997年,Mishra等[18]用原子球近似下的线性松饼-锡轨道方法(LMTO-ASA)和LDA计算未弛豫的Bi2Se3的带隙结构,得出其CBM和VBM都位于Γ点.Parker等[63]用GGA-PBE计算未弛豫的Bi2Se3的带隙结构表明CBM和VBM均位于Γ点,Bi2Se3为直接带隙半导体.但是,Larson等[25]利用LAPW和GGA计算未弛豫的Bi2Se3,得到的结果为间接带隙.此外,他们还对Bi2Se3进行了p1/2校正,发现了沿ZF方向的VBM符合六能谷模型.2012年,Luo等[64]报道,尽管晶格弛豫中包含vdW校正,但是带有GGA-PBE弛豫的Bi2Se3的VBM沿着ZF方向.
1975年,Köhler等[65]根据Bi2Se3的电磁学性质,首次提出了Bi2Se3为直接带隙半导体.ARPES的结果表明VBM位于Γ点[66].扫描隧道显微镜(STM)准粒子相互作用实验结果表明Bi2Se3为间接带隙半导体[67].
2012年,Yazyev等[67]利用LDA+G0W0的范数守恒赝势计算了Bi2Se3的能带结构.应用GW校正使VBM从ZF变到了Γ点[39].Nechaev等[66]通过G0W0计算和ARPES实验表明Bi2Se3为直接带隙半导体.他们指出,VBM只在有多体效应时才位于Γ点.Heremans等[54]使用LAPW和LDA+mBJ计算结果表明弛豫的Bi2Se3为直接带隙半导体,计算所得的费米面也与ARPES的结果一致[68-69].
综合以上实验结果可得,Bi2Se3与Bi2Te3相似的情况是,使用mBJ会减小在Γ点处的带隙,增大离Γ点较远处的带隙;与Bi2Te3不同的是,使用mBJ校正会对N型和P型Bi2Se3的Pisarenko曲线(Bi2Se3室温下塞贝克系数随载流子浓度变化的关系)产生影响,用mBJ校正计算所得的Pisarenko曲线与N型和P型Bi2Se3的实验数据吻合较好.使用LDA+mBJ的计算结果验证了Bi2Se3为直接带隙半导体.
3.2 Bi2Te3-xSex固溶体
实验表明,在Bi2Te3中用Se代替Te可以提高少子的热激发温度,因为Bi2Se3具有较大的带隙,为0.20~0.35 eV[70-71].1965年,Greenaway等[7]通过实验得出Bi2Te3-xSex中的x从0变化到1时,带隙值增大约2倍.然后随着x从1增加到3,带隙值逐渐减小.
无论计算时是否考虑GW或mBJ校正,得到的未弛豫Bi2Se3带隙均与实验值0.20~0.33 eV一致[70-71],如表2所示.但是,随着晶体结构的弛豫,带隙值减小.在考虑莫斯-布尔斯坦效应时,Bi2Se3的修正带隙低至0.16 eV[72].然而,使用ARPES得到的带隙为0.30 eV[73],这表明使用LDA+GW(或LDA+mBJ)计算时低估了带隙的值.所以Bi2Se3的带隙值仍然存在争议[74].
由于Bi2Te3-xSex样品大多存在富Te区,因此采用LDA计算Bi2Te3-xSex可以得到准确的结果.计算所得到的Bi2Se3和Bi2Te2.7Se0.3的能带结构表明,用Se代替Te增加了带隙值[75].

表2 Bi2Se3能带结构的计算方法、带隙类型和带隙值Tab.2 Calculated method,type of bandgap,and value of bandgap for Bi2Se3
4 Bi2-xSbxTe3的电子结构和电输运性能
近几十年来,Bi2-xSbxTe3合金一直是室温应用中最好的P型热电材料[76].已发现单晶Bi0.5Sb1.5Te3具有良好的室温热电性能,目前被广泛选择为基体材料[77-78].综合前沿报道,对Bi0.5Sb1.5Te3的能带特性进行了汇总和分析.
4.1 Sb2Te3二元化合物简介
Sb2Te3与Bi2Te3、Bi2Se3具有相似的能带结构和晶体结构,如图3.由于Sb2Te3具有层状结构,使其在电导率和热导率方面存在很大的各向异性.电导率和热导率在平行于c轴方向的值低于垂直于c轴方向的值.
2002年,Larson等[30]首先报道了未弛豫的Sb2Te3的能带结构,计算得到Nv=6[30].Wang等[31]用未弛豫晶格参数计算了Sb2Te3的能带结构,弛豫的Sb2Te3的VBM的简并度Nv=12,而未弛豫的Nv=6[31].此外,Sb2Te3能带结构的实验结果也存在争议.1967年,Schwartz等[79]测得Nv=6,而Middendorff等[80]测得Nv=12.

图3 Sb2Te3的晶体结构示意Fig.3 Crystal structureof Sb2Te3

与Bi2Te3相似的是,对于有mBJ校正的Sb2Te3,在Γ点附近的带隙减小,而在其他点的带隙增加.与Bi2Te3不同的是,不论是否采用mBJ校正,CBM的位置都保持不变[75].
4.2 Bi2-xSbxTe3固溶体
1988年,Stordeur等[83]的研究表明,对于 Bi2-xSbxTe3,通过热电输运性能和反射光谱得到的有效质量的态密度在x=1.5时达到最大值.Kim等[84]指出,在x=1.5取得,是由于2个不同价带的收敛[70],而不是价带展宽.ARPES结果表明在x=1.5处Bi2Te3和Sb2Te3的2个价带重叠[85].
根据计算结果,得出Sb2Te3的塞贝克系数低于Bi2-xSbxTe3的实验值[84,86-88],而Bi2-xSbxTe3的塞贝克系数与Bi2Te3的塞贝克系数相当,由于Bi2Te3基材料复杂的能带结构,测试结果可能存在一定的误差.Bi2-xSbxTe3的实测塞贝克系数仅比计算得出的Sb2Te3的塞贝克系数略高.所以,可以使用计算出的Sb2Te3的Pisarenko曲线(Sb2Te3室温下塞贝克系数随载流子浓度变化的关系)与Bi2-xSbxTe3的实验数据进行定性比较.
5 总结
主要介绍了(Bi1-xSbx)2(Te1-ySey)3热电材料的第一性原理计算的研究进展,就Bi2Te3、Bi2-xSbxTe3和Bi2Te3-xSex的晶体结构、能带结构及其影响能带结构的关键因素进行了分析.
根据Shubnikov-de Haas和de Haas-van Alphen的研究结果,六能谷模型已经被人们普遍接受.在利用实验晶格参数计算Bi2Te3的能带结构时,使用不同计算方法计算出的能带结构在间隙和能带上有很大差异,而结合SOI处理会使计算结果更为精确.通过分析Bi2Te3在范德华力作用下能带结构的变化,可以得到晶格参数的改变对Bi2Te3的能带结构影响很大.此外,Bi2Te3的周期势场会在受到应变作用时改变,进而影响材料的电子结构.而加压会使Bi2Te3材料的价带发生电子的拓扑转变,并导致Bi2Te3输运特性的反常.在考虑激发态电子计算能带结构时,使用GW或mBJ校正均可以使计算出的能带结构更精确.综合分析近年来的实验与计算结果,找到Bi2Te3的最优载流子浓度以及合理的改变其能带结构可以优化Bi2Te3材料的电输运性能.
mBJ或GW校正通过改变Γ点近和远点之间的能带极值能量差,对电子结构和电输运性能有很大影响.综合多种计算方法所得的结果,验证了Bi2Se3为直接带隙半导体.但是,不同的计算结果得到的Bi2Se3的带隙值差距太大,所以Bi2Se3的带隙值仍然存在争议.此外,Sb2Te3能带结构的计算结果与实验结果也存在争议.在单轴压力下,Sb2Te3基材料的VBM的会增大,从而优化热电性能.由于N型的Bi2Te3-xSex和P型Bi2-xSbxTe3固溶体大多存在富Te区,因此采用LDA进行计算可以得到准确的结果.根据比较,可以使用计算得出的Bi2Te3(Sb2Te3)的Pisarenko曲线来定性分析Bi2-xSbxTe3(Bi2-xSbxTe3)的实验结果,但是计算和实验之间仍存在一些偏差.
综上所述,因为晶体内部的缺陷会对载流子及声子的输运造成影响,从而影响热电性能,所以优化Bi2Te3基材料的热电性能必须要考虑实际样品的能带结构.因此,需要对具有不同缺陷的固溶体的能带结构进行定性研究.但是,由于Bi2Te3基材料具有复杂的能带结构,如何计算出准确的能带结构始终存在一定的争议.此外,受实验条件所限,范德华力以及应力也会对材料的能带结构造成一定的影响.因此,考虑到实际样品的能带结构与实验条件的不同,需要根据实验结果来定性分析材料的电输运性能.本综述总结了有关Bi2Te3基材料的能带结构的合理的计算方法以及影响能带结构计算的关键因素,为有效优化Bi2Te3基材料的热电性能提供了理论依据.
