基于分子模拟方法预测PIP2在双孔钾通道TREK-1上结合位点的研究
2021-08-16雷晓彤金怡卿孟烜宇
雷晓彤,金怡卿,孟烜宇
(苏州大学医学部放射医学与防护学院,定量生物与医学研究中心,放射医学与辐射防护国家重点实验室,江苏省高校放射医学协同创新中心,苏州 215123)
钾通道属于一类跨膜蛋白,在细胞膜上形成孔结构,控制细胞内外的钾离子传输.不同种类的钾通道在人体不同组织中分布广泛,与各种生理功能息息相关[1~3].钾通道的活性受到严格的调节,异常的通道活性与一系列疾病有关,如心血管疾病、癌症、癫痫及抑郁症等[4~9].因此钾通道是药物研究的重要靶标之一[10~13].
钾通道根据跨膜区域拓扑结构的区别,可以分成3类:具有两个跨膜片段和一个孔道结构域的内向整流钾通道(Kir)[14],由6个跨膜片段和一个孔道结构域组成的电压门控钾通道(Kv)[15]以及包括4个跨膜片段和两个孔道的双孔钾通道(K2P)[16].K2P通道是二聚体结构,每个亚基有4个跨膜结构域M1~M4,两个孔结构域P1和P2以及位于胞内的N端和C端[17](图1).迄今为止,已发现有15个K2P通道成员,分属6个亚家族,即弱内向整流串联孔域钾通道(TWIK)、串联孔域氟烷抑制钾通道(THIK)、与TWIK相关钾通道(TREK)、TWIK相关的花生四烯酸敏感钾通道(TRAAK)、与TWIK相关酸敏感钾通道(TASK)、TWIK相关碱性激活钾通道(TALK)以及与TWIK相关的脊髓钾通道(TRESK)[18,19].
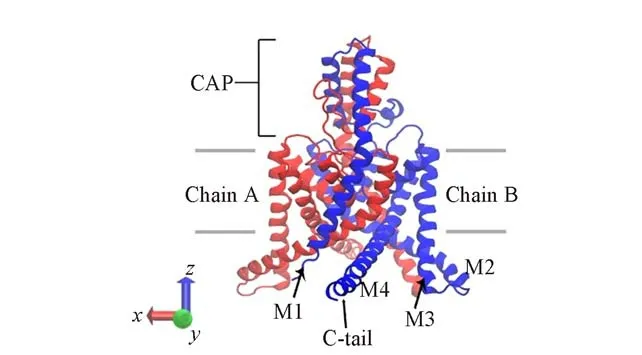
Fig.1 Structure of channel protein TREK-1The structure of TREK-1 is shown in ribbons with chains A and B in red and blue,respectively.Key structural elements are marked in the figure.
哺乳动物K2P2.1通道(TREK-1,或称为KCNK2)广泛分布于神经系统和心脏中,在神经保护、麻醉、痛感和抑郁等方面的细胞机制中起关键作用[19~21],是两孔结构的钾通道类别的重要成员.TREK-1可在静息电位时处于开放态,因此被称为背景钾通道或漏流钾通道.其活性受磷脂、花生四烯酸、神经递质、胞内pH值、机械拉伸和温度等多种因素的调节[22],许多临床药物也会影响TREK-1通道的活性,如抗精神病药氯丙嗪[23]和抗抑郁药氟西汀[24].
磷脂酰肌醇4,5-二磷酸酯(PIP2)是分布在细胞质膜内层中的信号脂质分子,仅在膜的内层分布,约占细胞膜总磷脂含量的1%,但行使的生物学功能却十分复杂[25,26].除了公认的PIP2的水解产物发挥的双信使作用之外[27~29],PIP2还与多种离子通道、转运蛋白等相互作用,并调节这些蛋白的功能.PIP2与钾通道相互作用的研究是探索离子通道门控机制的重要研究内容之一[30~36].如PIP2为Kir通道的重要激动剂[37,38];PIP2控制三磷酸腺苷敏感性钾通道(KATP)的ATP敏感性并促进通道开放[39];PIP2还参与调节Kv通道功能等[40].
PIP2可调节TREK-1通道的活性[41~43].电生理实验表明,PIP2对TREK-1通道具有浓度依赖的双向调节作用:PIP2高浓度对通道起抑制作用,PIP2降低部分浓度后激活通道,PIP2浓度耗尽后抑制通道[44];TREK-1通道的羧基末端结构域(即M4螺旋的碳末端)对通道与PIP2的相互作用有重要影响.TREK-1晶体结构的解出为在分子层面上研究TREK通道与PIP2的相互作用提供了条件.本文将主要基于分子对接(Docking)和分子动力学(Molecular dynamics,MD)模拟对PIP2在TREK-1通道上的可能结合位点进行预测,并探索了TREK-1与PIP2结合的分子特征.
1 计算方法
1.1 分子对接
使用分子对接程序AutoDock 4[45]将PIP2对接到TREK-1通道蛋白(PDB ID:6CQ6)[46]的晶体结构中.由于PIP2分子的两条脂肪长链具有较多的自由度,给柔性对接带来困难,因此在对接中将其替换为类似物diC1,其用两个甲基分别取代PIP2的两个末端脂肪链(图2).我们曾用同样的策略研究了PIP2与Kir,小电导钙激活钾通道(SK)等通道的相互作用[47,48].对接中,使用C,H,N,O,P元素在均匀的网格上采样得到TREK-1通道蛋白的网格电势图,网格包含49×107×41个点,点间相距0.0375 nm,中心坐标设为(43.588,17.002,-8.652).选择Lamarckian遗传算法生成配体与蛋白的结合构象.将TREK-1中亚基B上的K45,R207,R297,K301,K302和K304的侧链设为柔性.进行了100次对接模拟,此后根据diC1与TREK-1对接的结合能以及diC1与膜的相对空间取向选择可能的复合物构象.
1.2 分子动力学模拟
首先,将对接得到的diC1构象加上两条脂肪链,恢复完整的PIP2结构,并用CHARMM力场进行结构优化,得到PIP2与TREK-1复合物结构.将复合物嵌入1-棕榈酰-2-油酰-卵磷脂(POPC)磷脂膜,膜的尺寸为10.0 nm×10.0 nm.将通道蛋白、小分子和膜溶解在10.0 nm×10.0 nm×15.0 nm的水盒子中,添加0.15 mol/L KCl溶液以模拟生理盐水,并使模拟系统总电荷为零,由CHARMM-GUI[49]完成.另外建立了对照组,只包括通道蛋白与POPC膜,去除PIP2,其余条件均相同.
采用GROMACS软件包[50]进行MD模拟.使用VMD软件[51]可视化模拟结果.TREK-1和PIP2使用CHARMM36力场[52],溶剂水分子使用TIP3P水模型[53].使用V-rescale算法将温度保持在298 K[54].在所有方向上应用周期性边界条件.使用PME方法处理长程静电相互作用,并以1.2 nm的截断距离计算范德华相互作用[55,56].使用LINCS算法保持水分子键长.模拟中,步长为2.0 fs,每2 ps保存一次数据.每个体系进行30 ns的模拟,以充分优化复合物的结构.
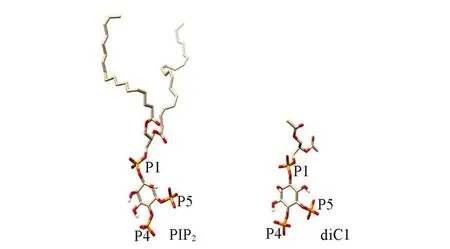
Fig.2 Structures of PIP2 and diC1The structures of PIP2 and its analog diC1 used in molecular docking,with carbon in golden,phosphorus in orange,oxygen in red,and hydrogen in white(only polarized hydrogen atoms are shown).
1.3 均力势计算
均力势(PMF)计算使用伞型抽样[57~59]方法,沿膜的法线方向(-z方向,图1)拉动PIP2,由此计算PIP2和TREK-1通道结合的PMF值.PIP2到通道蛋白的距离(d)用谐波力(F)限制在参考距离(d0):F=k×(d-d0),其中力常数k=2000 kJ·mol-1·nm-2.采样窗口间隔为0.1 nm,拉伸距离是1.5 nm,一共设置了15个采样窗口.平衡后每个窗口进行3 ns模拟.PMF曲线用g_wham工具获得[60~62],由此得到两个可能的结合位点上PIP2与通道蛋白TREK-1的自由能曲线并进行比较.
2 结果与讨论
2.1 对接结果
PIP2的肌醇头部有两个磷酸根,在生理pH下通常带有3~4个负电荷[63],能与受体蛋白形成静电吸引.之前对PIP2与内向整流型钾通道和电压门控钾通道相互作用的研究也表明,PIP2结合在这些通道的正电富集的区域,与通道上的数个碱性残基形成稳定的盐桥[47,64].因此,计算了TREK-1通道的表面静电势(图3).在TREK-1的位于膜内侧的膜水交界处存在正电区域,一方面,膜内侧的膜水交界处的位置为PIP2肌醇头部能够直接接触的蛋白位置;另一方面,这个区域包含有碱性残基K45,R207,R297,K301,K303,K304及R311等,具有与PIP2头部磷酸根形成盐桥的可能性,因而这些正电区域是潜在的PIP2的结合位点.
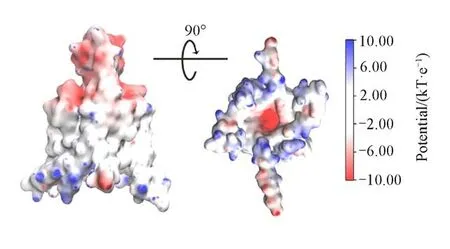
Fig.3 Electrostatic surface of TREK-1 calculated using APBS method
TREK-1通道由两个同源亚基A和B构成,沿膜结构中结构较为完整的亚基B与diC1进行对接.对接共进行100次模拟,产生100个构象,统计这100个构象中diC1在TREK-1的分布情况,取diC1小分子每个构象的P1原子的位置信息,获得diC1与TREK-1结合的簇(图4).可见,对接的diC1主要集中在通道的两个区域,位点A:位于亚基B的M4螺旋与M1螺旋之间(红点标示);位点B:位于B链M4螺旋与M3螺旋之间(绿点标示).的法线方向,即z方向,具有二重对称性,选择晶体

Fig.4 One hundred sampled positions of PIP2 onto the TREK-1 channelP1 atomic coordinate of PIP2 in each sampled conformation is marked with spot.A sampled position in the A site where is between the M1 and M4 helices is indicated by a red spot;A sampled position in the B site where is between the M3 and M4 helix is indicated by a green spot.
这两个位点都包含数个能与diC1的头部磷酸根成静电吸引的正电残基,从而稳定diC1与通道的相互作用.根据计算出的diC1与通道蛋白的结合能,以及其在这两个位点的空间位置选择可能的构象.从位点A的数个对接构象中选择第36个构象,Autodock估算的结合能为-53.33 kJ/mol[图S1(A),见本文支持信息].这个构象中,diC1的P1原子键连的O原子与TREK-1通道B链的M4螺旋上的K304形成盐桥,P4键连的O原子会与同样位于M4螺旋上的R297和K301形成盐桥,P5键连的O原子与M1螺旋上的K45形成盐桥.
位点B的对接构象中选择了第80个构象,Autodock估算的结合能为-33.40 kJ/mol[图S1(B)].这个构象中,diC1的P1键连的O原子会与TREK-1通道B链的M4螺旋上的K302成盐桥,P4键连的O原子和P5键连的O原子与M3螺旋上的R207残基接触.对位点A和位点B的两个构象的diC1加入两条脂肪链,形成完整的PIP2分子,再经过能量最小化,获得进一步MD模拟的构象.
2.2 分子动力学模拟结果
分别对从对接结果中选择出来的两个位点的构象进行全原子MD模拟.MD模拟可以充分考虑受体和配体的柔性,进一步优化构象,探究两个可能结合位点上PIP2与TREK-1通道相互作用的具体情况.图S2(见本文支持信息)为使用CHARMM-GUI生成的模拟体系.分别模拟了两组PIP2位于位点A(Site A)上(A1,A2),两组PIP2位于位点B(Site B)上(B1,B2)以及只含有通道不含PIP2的对照组(Control).每个体系在1 ns的系统平衡之后均进行30 ns的正式模拟(Production run).
2.2.1 位点A构象与TREK-1通道蛋白的相互作用 位点A中,PIP2位于TREK-1通道B链的M4螺旋与M1螺旋之间的空隙中.经过30 ns的模拟,PIP2基本稳定在这个位点.取A1组作为代表轨迹进行分析,TREK-1通道的均方根误差(RMSD)在10 ns之后稳定在0.4 nm左右(图S3,见本文支持信息).与初始构象[图5(A)]相比,从模拟轨迹的最终构象[图5(B)]可见,PIP2向通道B链M4螺旋的碳末端稍稍移动.以PIP2的初始位置和构象(即对接的构象)为参考结构,计算30 ns轨迹中PIP2整个分子的RMSD[图S4(A),见本文支持信息],在模拟16 ns后,整个分子的RMSD稳定在了0.7 nm左右.因此,相较于初始位置,在一段时间的MD之后,PIP2发生了一些位移.考虑到PIP2头部对与通道结合的重要性和敏感性,还计算了PIP2头部的RMSD,同样参考头部的初始位置,计算结果和整个分子的RMSD变化呈现相同趋势,在16 ns之后稳定在0.8 nm左右[图S4(B)].

Fig.5 Initial conformation in the MD simulation at the A site(A)and the final conformation after 30 ns simulations(B)
图5 (B)给出了经过MD模拟后PIP2和TREK-1具体的相互作用情况.粗棒显示出与PIP2距离0.5 nm之内的TREK-1上的氨基酸残基.PIP2的P1原子分别与M4螺旋的K304和M1螺旋的K45结合,P4原子与M4螺旋的R311接触,P5原子分别与M4螺旋的R311和M1螺旋的K45紧密结合.与初始对接的P1-K304,P4-R297/K301,P5-K45有所区别.PIP2失去了与M4的R297和K301的盐桥,同时形成了在M4上的与R311的盐桥.R297和K301侧链位于同一螺旋的一侧,在对接中将其设为柔性以便与diC1产生相互作用;在MD中,考虑到静电排斥,这两个末端带有正电的侧链从同一侧同时与PIP2相互作用可能并不稳定,最终两个侧链改变了方向不再与PIP2相互作用.PIP2转而与位于M4靠近碳端的R311建立盐桥.考虑到盐桥对稳定PIP2与通道结合的重要性,计算了整个MD轨迹中PIP2分别与K45,R297,K301,K304,R311最短距离的变化情况.如图S5(A)(本文支持信息)所示,在MD最后一帧的构象中PIP2与K45,K304和R311形成的盐桥在轨迹中均稳定存在.
除了盐桥相互作用,0.5 nm之内与PIP2头部有接触的残基有T303和V307,均位于M4螺旋.由此可见,带有净负电荷的PIP2与通道蛋白的正电残基之间的静电相互作用起到了稳定两者结合的主要作用.另外,在模拟过程中,PIP2的两条脂质链不断运动以寻找更好的伸展方式,与TREK-1通道跨膜段上的部分疏水氨基酸形成相互作用,如L52,V53,V55,L56及I60等,这些氨基酸都是位于通道B链的M1螺旋上,以疏水相互作用稳定PIP2与TREK-1通道蛋白的结合.总之,在整个模拟过程中,PIP2在TREK-1通道的A位点发生了小的位移,但并未脱离位点,PIP2结合在此位置具有可能性.A2组也取最后一帧进行构象分析,如图S6(A)(本文支持信息)所示,结合模式与A1组相似.
基于模拟16 ns后的动力学轨迹计算PIP2与TREK-1通道的相互作用能.总的相互作用能为(-616.80±96.71)kJ/mol,其中静电相互作用能为(-544.36±91.61)kJ/mol,范德华相互作用能为(-72.44±20.62)kJ/mol.静电相互作用能占据明显优势,再次说明PIP2的头部基团与TREK-1蛋白的正电残基的相互作用对稳定整个复合体构象具有重要作用.
2.2.2 位点B构象与TREK-1通道蛋白的相互作用 对位点B进行了与位点A相同的MD模拟,观察PIP2与通道在此位点的相互作用情况.两组轨迹B1和B2的最后构象基本一致[图6(B)和图S6(B)],以B1作为代表轨迹进行分析.PIP2位于通道B链的M3与M4螺旋之间,经过30 ns模拟后,通道的RMSD稳定在0.3 nm左右(图S3).模拟刚开始时,PIP2在空间上偏向M3螺旋,如P4,P5原子都与M3螺旋的R207接触;随着模拟时间的进行,相比初始构象[图6(A)],整个PIP2分子逐渐地靠近M4螺旋,但仍然与M3有直接相互作用,整体位置保持在位点B之中.从PIP2的RMSD可见,同样参考PIP2的初始位置和构象,模拟16 ns后整个分子的RMSD稳定在0.5 nm左右[图S4(A)];PIP2头部的RMSD也在模拟16 ns后保持在0.5 nm左右[图S4(B)].比位点A的PIP2的RMSD小0.2~0.3 nm,位点B中的PIP2在MD过程中位移更小.
从图6(B)可见,模拟最终构象的M3和M4螺旋上的一些残基与PIP2均有接触.并统计了距离PIP2分子0.5 nm之内的通道蛋白的氨基酸.其中,PIP2的P1原子与M4螺旋的K302接触,P4原子与M4螺旋的R297形成盐桥,P5原子与M3螺旋的R207形成盐桥.与对接得到的盐桥作用网络有细微差别:在MD之后,P4失去原先与M3上的R207的盐桥,转而与M4上的R297形成盐桥.计算了PIP2与R207,R297,K302在整个轨迹中最短距离的变化情况[图S5(B)],R297在模拟23 ns后与PIP2形成盐桥,R207和K302与PIP2的盐桥在整个轨迹中都很稳定.除碱性残基外,一部分非极性氨基酸,如A链的F158,I160,I161,L165,B链的V64,I292,W295,V298,I299等,与PIP2的两条脂肪链产生疏水作用,稳定了两者的结合.

Fig.6 Initial conformation in the MD simulation at the B site(A)and the final conformation after 30 ns simulations(B)
基于模拟16 ns后的动力学轨迹计算PIP2与TREK-1通道的相互作用能.总的相互作用能为(-628.73±49.16)kJ/mol,其中静电相互作用能为(-477.64±61.50)kJ/mol,范德华相互作用能为(-151.09±21.01)kJ/mol.与位点A的情形类似,静电相互作用能占据明显优势.PIP2在两个结合位点的总相互作用能处于同一个水平;位点A中PIP2与通道蛋白的静电作用能大于位点B(部分由于A中更多的盐桥作用);而位点B中PIP2与通道的范德华相互作用能大于位点A(涉及B中更多的PIP2脂肪链与跨膜螺旋的疏水作用).
从PIP2在A和B两个位点的结合结果可见,位点A经过MD模拟后与PIP2形成稳定盐桥的正电残基有M1:K45,M4:K304/R311;B位点与PIP2形成稳定盐桥的正电残基有M3:R207,M4:R297/K302.两个位点中,富含正电残基的M4螺旋都贡献了稳定盐桥.Chemin等[41]认为M4螺旋上的5个正电残基与PIP2对通道的正向激活作用有关;这5个正电残基分别对应本结构中的R297,K301,K302,K304和R311.与预测结果相符:K304和R311在位点A中与PIP2形成盐桥;而R297和K302在位点B中与PIP2形成盐桥.此外,PIP2与TREK-1的相互作用较为复杂.PIP2低浓度时激活通道而高浓度时抑制通道活性[44].Woo等[18]的研究表明,M4螺旋的位于5个正电残基下游的3个连续精氨酸与PIP2对通道的抑制作用有关.由于这一部分的结构在TREK-1的晶体结构中缺失,并未包含PIP2抑制通道的结合位点.
2.2.3 均力势计算 选取模拟体系30 ns的最终构象,沿膜的法线方向(-z方向,图1)向外拉PIP2,以伞状采样方法计算PMF,分别估算PIP2在两个位点与TREK-1的结合自由能,结果如图7所示.可见,A位点的结合自由能为-165.16 kJ/mol;B位点的自由能为-41.02 kJ/mol.两个位点PIP2的结合自由能具有显著差异.由PMF计算的结合自由能与由Autodock打分函数给出的初始构象的结合自由能大小一致.从能量角度来说,两个位点结合PIP2能力有差别,PIP2更倾向与TREK-1通道蛋白上的A位点结合.
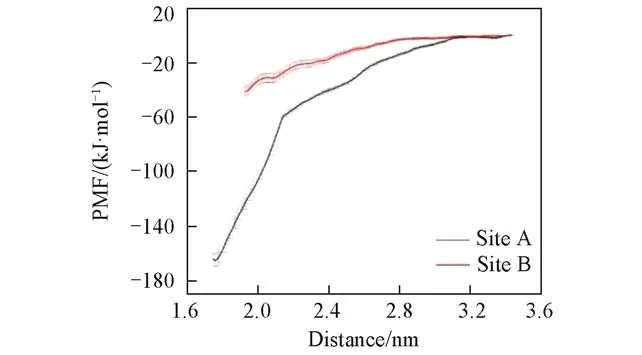
Fig.7 PMF values of the two binding sites calculated by the umbrella sampling method
3 结 论
利用分子对接和分子动力学模拟(MD)方法研究了磷脂酰肌醇4,5-二磷酸(PIP2)在双孔钾通道TREK-1上可能的结合位点和相互作用.Autodock对接结果显示,PIP2在TREK-1通道上的结合位点可能有两处:(1)B链M4螺旋与M1螺旋之间(位点A);(2)B链M4螺旋与M3螺旋之间(位点B).在可能的两种结合位点上分别选取了代表构象,并进行MD模拟.MD模拟能够充分考虑受体-配体的柔性,从而优化对接构象.在30 ns的模拟时间内,PIP2在两个位点中均未脱离位点,在位点内有一定程度的位移和构象变化.经过MD模拟后,位点A中与PIP2形成稳定盐桥的正电残基有M1:K45,M4:K304/R311;B位点中与PIP2形成稳定盐桥的正电残基有M3:R207,M4:R297/K302.两个位点中,富含正电残基的M4螺旋都贡献了稳定盐桥.从结构上来说,M4螺旋的两侧都有空间可以容纳PIP2.M4与一侧的M1共同构成位点A,与另一侧的M3共同构成位点B.PMF计算显示,PIP2在位点A与TREK-1的结合自由能高于在位点B.从能量角度来说,PIP2将优先在位点A与TREK-1结合.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20210106.
