过渡金属改性的Cr-Ce-O催化剂氧化氯苯性能
2021-08-09徐功达赵云霞陈敏东南京信息工程大学环境科学与工程学院江苏省大气环境与装备技术协同创新中心江苏省大气环境监测与污染控制高技术研究实验室江苏南京20044南京信息工程大学化学与材料学院江苏南京20044
徐功达,司 涵,黄 琼*,陶 涛,2,杨 波,赵云霞,陈敏东 (.南京信息工程大学环境科学与工程学院,江苏省大气环境与装备技术协同创新中心,江苏省大气环境监测与污染控制高技术研究实验室,江苏 南京 20044;2.南京信息工程大学化学与材料学院,江苏 南京 20044)
含氯挥发性有机化合物(CVOCs)因具有化学性质稳定、热稳定性好和不易被生物降解等特点,在环境中长期累积存在,且具有较高的毒性和致癌、致畸作用,是一类严重威胁人类和生物健康、破坏自然生态环境的有机物[1].CVOCs的净化治理技术主要包括焚烧法、冷凝法、生物法、催化燃烧法等,其中催化燃烧技术适用于低浓度废气的处理,具有低能耗、效率高以及无二次污染物等优点,广泛应用于实际生产中.催化氧化消除方法是将烟气中的CVOCs直接分解转化为无毒或毒性小的 H2O、CO2和HCl/Cl2[2].然而在催化氧化CVOCs反应过程中所产生的HCl/Cl2极易促使催化剂中毒而失活,影响其消除效果[3].
目前,催化氧化 CVOCs催化剂主要包括贵金属、非贵金属和分子筛型催化剂3类.贵金属催化剂主要以Pt、Pd、Rh等贵金属为活性组分,但价格昂贵,需高表面积的载体担载高分散的贵金属颗粒,且遇Cl易中毒[4],副产物的毒性大于反应物本身;非贵金属催化剂主要有过渡、稀土金属氧化物和钙钛矿型复合氧化物催化剂等,但氧化活性整体仍不及贵金属催化剂.分子筛型催化剂因具有较好的吸附性能和较大的结构灵活性[5-6],适宜的B酸位和L酸位而有助于催化氧化性能的发挥和抗氯中毒性能的提高,分子筛催化剂还可以通过金属或金属氧化物改性,得到氧化活性更佳的改性分子筛,因此研究学者对改性分子筛型催化剂进行广泛研究,利用浸渍法、离子交换法、共沉淀法和物理气相沉积法等制备的Cu、Co等金属改性的分子筛型催化剂,催化氧化活性不尽相同[7].
由于CeO2晶胞中存在Ce3+和Ce4+2种离子的氧化还原循环且具有良好的储氧性能,因而在催化氧化领域具有广泛的应用[8].Lu等[9]采用溶胶-凝胶柠檬酸法制备了一系列高效的MnOx-Co3O4- CeO2三效催化剂,当物质的量比为 16:19:1,反应温度为100℃时,催化剂对 HCHO 的去除效率达到最高.José等[10]采用沉淀法制备 CexZr1-xO2(x=0,0.15,0.5,0.8,1.0)催化剂催化氧化1,2-二氯乙烷和三氯乙烯,研究发现,混合氧化物催化剂相对于纯氧化物显示出更高的氧化活性,且不同Ce/Zr物质的量比将深刻影响催化剂氧化活性,其中 Ce0.5Zr0.5O2活性最佳.梁文俊等[11]采用浸渍法在 0.5% Pd/Al2O3催化剂上负载质量分数为 1%、2%、2.5%、3%、4%的 CeO2,随着CeO2负载量的增加,催化剂催化氧化氯苯的活性先升高后降低,当 CeO2负载量为 2.5wt%时,催化剂展示出最佳的氧化活性.Wang等[12]通过引入MnOx,由溶胶-凝胶法制得的 CeOx-MnOx复合金属氧化物催化剂可显著提高其催化活性和稳定性,催化活性最佳的 CeO2-MnOx(0.86)催化剂在 254℃可将氯苯完全氧化为HCl、Cl2、CO2和微量CO,且无多氯苯形成.
由于过渡金属氧化物,如MnOx,CuO,CoOx等,在催化氧化过程中均会显现出不同程度的氯中毒失活或产生大量含氯副产物等问题,因此提高氯的抗性和抑制副产物的生成是催化降解氯挥发性有机物(CVOCs)的关键问题.Feng等[13]合成了一种卵黄壳型介孔钴氧化物催化剂,研究表明所制催化剂中Co与Cr元素间的强相互作用能有效促进活性Cr6+生成,且具有良好的还原性和活性氧物种(O2-,O-和Oads2-)迁移率,有效地提高了催化活性(T90=325℃).Zhang等[14]分别采用聚甲基丙烯酸甲酯(PMMA)模板法和初湿浸渍法制备了三维有序大孔(3DOM)CeO2、3DOM Cr2O3、3DOMxCr2O3-CeO2(x为 Cr2O3的重量百分比=3.5,5.5,8.0wt%)和 5.5wt% Cr2O3/3DOM CeO2样品.结果表明,3DOM5.5Cr2O3-CeO2的良好催化性能与其负载浓度、低温还原性和Cr2O3之间的强相互作用有关.氧化铬在催化氧化CVOCs过程中表现出较好的抗氯中毒性能,且性能稳定,且副产物产生较少.虽然改性后的铬基、铈基催化剂已经应用于CVOCs的去除,但二者的结合以及载体类型、微观结构、活性组分负载量等方面仍有待进一步改进以提高催化剂氧化性能及抗 Cl中毒能力.为此,本文以混合氧化物Cr2O3和CeO2为活性组分基底,以Al2O3,堇青石和ZSM-5为载体,采用柠檬酸络合法制备负载型 Cr-Ce-O催化剂,探索Cr/Ce物质的量比、过渡金属氧化物掺杂、载体、掺杂量以及焙烧温度对催化氧化氯苯性能的影响,并利用XRD、BET、XPS、H2-TPR、SEM等分析手段表征催化剂结构的组成、粒径以及形貌,以确定制备方法与催化剂氧化活性间的内在关系,优化设计出热稳定性好、比表面积大、氧化能力强,且具有良好的抗氯中毒性能的催化剂.
1 材料与方法
1.1 催化剂的制备
催化剂采用柠檬酸络合法制备,取 6.9g硝酸铈(Ce(NO3)3·6H2O)和 6.4g 硝酸铬(Cr(NO3)3·9H2O) 分别加入 20mL去离子水中,充分搅拌溶解,再向上述溶液中加入 1.0g一水合柠檬酸,继续搅拌待完全溶解,溶液经磁力搅拌并加热直至形成溶胶状,将 5.0g条形 Al2O3载体浸渍其中 2h,取出后先 80℃,干燥12h,再在马弗炉中500℃焙烧4h,取出后经方孔筛网过筛去除多余粉末,制得Cr/Ce物质的量比为1:1的Cr2O3-CeO2/Al2O3催化剂,其金属氧化负载量为20wt%,柠檬酸与金属离子物质的量比为1:6.为进行比较,同时制备Cr/Ce物质的量比为4:1,3:1, 2:1,1:2,1:3和1:4;以及掺杂有NiO,MnOx,CoOx,CuO,FeOx和ZrO2的Cr2O3-CeO2/Al2O3催化剂,负载量为20%;负载于堇青石、ZSM-5载体和不同焙烧温度的Cr-Ce-O催化剂均采用上述制备方法.
1.2 催化剂的表征
实验采用X射线多晶衍射仪(XRD, XRD-6100)对样品进行物相结构检测,使用 Cu Kα辐射(λ=0.15406nm),扫描范围为 2θ = 10~80°.扫描电镜(SEM,HITACHI SU1510)分析样品的形貌及微观结构等,工作电压为15kV.使用ASAP 2460自动气体吸附分析仪测量催化剂样品的比表面积 SBET和孔容孔径,通过BET方程计算样品比表面积SBET,通过BJH方程计算孔容(V).用 X射线光电子能谱(KRATOS AXIS ULTRA(DLD) )表征催化剂表面元素含量和其存在形态,测定混合氧化物中Cr、Ce和O等原子的结合能.利用AutoChem II 2920吸附仪和TCD检测器测定氢气程序升温还原(H2-TPR),用以描述材料的氧化还原能力.
1.3 催化活性实验
催化剂氧化性能测试在连续流固定床中进行,石英管内径为 80mm.在反应器装入 4.0g催化剂,进料气体流量为 800mL/min,体积空速(GHSV)保持在10000h−1.反应器内的混合气体由空气和氯苯混合而成,氯苯浓度由质量流量控制器保持进气流速为500×10-6,反应器的温度由固定床外围管式炉中的热电偶控制.混合气体的组成采用在线气相色谱仪分析,其配备了 Restek Rtx-1(0.25µm×0.25μm×30m)火焰离子化检测器(FID).氯苯的转化率根据氯苯的初始浓度和反应后浓度计算得到.

2 结果与讨论
2.1 Cr-Ce-O催化剂的性能测试
2.1.1 Cr/Ce物质的量比对Cr2O3-CeO2/Al2O3催化剂氧化性能的影响 由图 1可知,随着反应温度的升高,催化剂对氯苯的催化转化率逐渐升高,且不同Cr/Ce物质的量比的Cr2O3-CeO2催化剂对氯苯的催化转化效率不尽相同.当 Cr/Ce物质的量比 1:2时,其催化活性优于其它催化剂,其结果与比表面积结果相一致,但催化氧化氯苯转化率在反应温度500℃时仍仅为 84.7%,催化氧化效率低且反应温度高,未能达到理想催化剂性能,因此实验以Cr/Ce物质的量比为 1:2为基础,进一步对 Cr2O3-CeO2催化剂进行深入优化改进.

图1 不同Cr/Ce物质的量比的Cr2O3-CeO2/Al2O3催化剂催化氧化活性Fig.1 The catalytic activity of Cr2O3-CeO2/Al2O3 catalysts with the different molar ratios of Cr/Ce
2.1.2 过渡金属氧化物掺杂对 Cr2O3-CeO2/Al2O3催化剂氧化性能的影响 部分过渡金属氧化物在催化剂催化氧化CVOCs过程中亦表现出对氯良好的抵抗能力[15].由图 2可知,掺杂有不同过渡金属氧化物的催化剂氧化活性不同,具体表现为 ZrO2>NiO>MnOx>Co2O3>FeOx>CuO,其中掺杂有 ZrO2的Cr2O3-CeO2/Al2O3催化剂表现最佳,反应温度从300℃升高至 350℃时,催化剂活性提升最为明显,当反应温度为 350℃时,催化剂较未经任何掺杂的Cr2O3-CeO2/Al2O3催化剂相比其氧化活性提高了23.9%.

图2 不同过渡金属掺杂的Cr2O3-CeO2/Al2O3催化剂氧化活性Fig.2 The catalytic activity of Cr2O3-CeO2/Al2O3 catalyst doped with different transition metals
2.1.3 载体材料对Cr2O3-CeO2(Cr/Ce=0.5)催化剂氧化性能的影响 掺杂改性后的催化剂催化活性得到一定程度的提升,但均未达到理想效果,催化转化率大于90%所需温度仍高于450℃以上,其原因可能是载体Al2O3长时间暴露在高温和高湿环境中,逐渐烧结并从γ相转变为α相[16],表面积和热稳定性降低所致.对于过渡金属氧化物催化剂,其催化活性不仅取决于活性组分氧化还原性能以及酸性,其载体的性质在提高负载物活性和耐久性方面也起着重要作用[17].选取堇青石、ZSM-5 2种不同载体与Al2O3相比较,探讨其对催化剂氧化活性的影响,其结果如图3所示.
由图3可知,Al2O3与堇青石负载的催化剂整体催化活性亦随反应温度的升高而显著提高,且优于ZSM-5,而 Cr2O3-CeO2/ZSM-5催化剂在反应温度300~400℃区间时,其对氯苯催化氧化性能的提升较为缓慢.继续升高反应温度至 500℃时,氯苯转化率有所提升,但催化剂整体氧化性能劣于以Al2O3与堇青石为载体的催化剂.研究发现,以堇青石为载体的催化剂氧化活性最佳,当反应温度为 450℃时,催化氧化氯苯转化率可达97.8%.

图3 不同载体负载的Cr2O3-CeO2催化剂催化活性Fig.3 The catalytic activity of Cr2O3-CeO2 catalysts supported on different supports
2.1.4 过渡金属掺杂量对 Cr2O3-CeO2/堇青石催化剂催化活性的影响 一般来言活性组分越多,提供的活性位越多,越有利于反应的进行,但负载量并非越多越好,当活性组分负载量超过分散阈值后,多余晶相会覆盖活性位以及堵塞催化剂表面孔道,阻碍氧化反应的进行致使催化剂氧化活性下降[18].为研究金属掺杂量对催化剂性能的影响,本文以具有最佳性能的堇青石为载体,掺杂有 ZrO2和 NiO的Cr2O3-CeO2为活性组分,制备 Cr2O3-CeO2-MxO(M=Ni、Zr,x为M/Cr物质的量比,x= 0.25, 0.50和0.75)/堇青石催化剂,其氧化活性如图4所示.
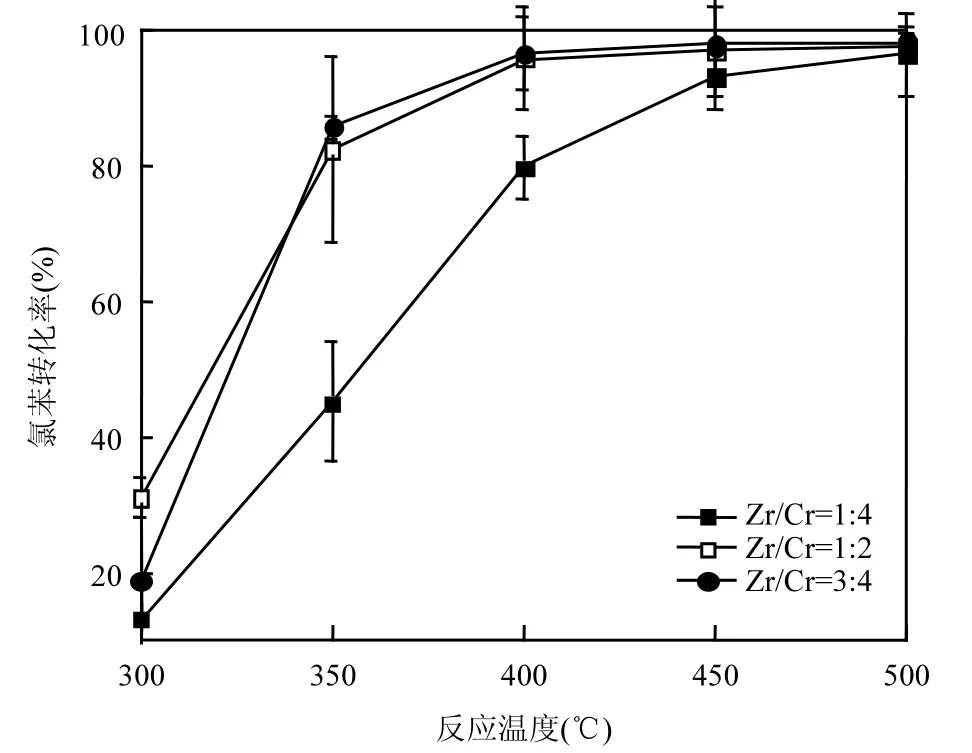
图4 不同掺杂量的Cr2O3-CeO2-MOx/堇青石催化剂催化活性Fig.4 The catalytic activity of Cr2O3-CeO2-MOx/cordierite catalysts doped with different amounts
由图 4可知,掺杂量对催化剂的氧化活性产生显著性影响.当Zr/Cr物质的量比分别为1:4时,反应温度为400℃时的催化氧化转化率仅为80.0%,而当Zr/Cr物质的量比为1:2和3:4时,催化剂的氧化活性显著提高,反应温度为350℃时的催化氧化转化率达到 85.8%,两者氧化活性相似,无显著性差别.因此研究将以Zr/Cr物质的量比为1:2为基础,进一步设法通过微观结构调控提升催化剂氧化性能.
2.1.5 焙烧温度对催化剂氧化性能的影响及稳定性 考虑到不同焙烧温度对催化剂表面结构和孔道结构产生重要影响,选取所制备活性最佳的催化剂为研究对象,即 Cr2O3-CeO2-ZrO2/堇青石(Cr/Ce=1:2,Zr/Cr=1:2),设置焙烧温度分别为 300,400和500℃,焙烧时间4h.
由图 5可知,焙烧 4h后,随着焙烧温度由300℃升高至 400℃时,所制催化剂氧化活性得到显著提高,反应温度为 350℃时催化转化率可达92.0%,其CO2的选择性高达87.0%;而随着焙烧温度进一步提升至500℃时,催化氧化氯苯转化率反而降低.因此认为催化剂最佳焙烧温度为 400℃,其与催化剂表面结构和介孔结构有关,当焙烧温度为 400℃时,催化剂表面较 300℃焙烧催化剂更为粗糙,显现出更多的活性位点,而当焙烧温度升高至 500℃,催化剂呈现大量大孔结构,发生熔融现象,比表面积降低.

图5 焙烧温度对催化剂催化氧化氯苯的影响Fig.5 Effect of calcination temperature on catalytic oxidation of Chlorobenzene
考察该催化剂的稳定性.催化剂在350℃持续催化氧化氯苯,每15min记录数据1次,合计进行24h.发现催化剂催化氧化氯苯转化率始终保持在99.0%以上,无明显催化剂中毒现象的发生.
2.2 催化剂的表征结果
2.2.1 催化剂的结构 由图 6(a)可知,在 28.5°、47.5°和 56.3°处可明显观察到属于 CeO2立方萤石结构的特征衍射峰(JCPDS #34-0394),在 33.6°、36.2°、41.5°、44.2°、54.9°、58.4°可观察到属于 Cr2O3尖晶石结构的特征衍射峰(JCPDS, PDF 6-504),且发现Cr的衍射峰较小,说明有部分Cr进入到Ce晶格中并与之形成CrCeOx固溶体[19],位于28.5°处的衍射峰随着Ce含量的增大,其衍射峰强度呈现出先增大后减小的趋势,Cr/Ce物质的量比为1:2时峰更窄而且明显,这说明Cr/Ce物质的量比为1:2时,产生的CrCeOx固溶体晶型更完整[20-21].如图 6(b),不同过渡金属氧化物掺杂的Cr2O3-CeO2/ Al2O3,各催化剂上均出现 CeO2、Cr2O3和掺杂相对应的金属氧化物的特征谱峰,且与图 6(a)相比,掺杂ZrO2后的CeO2衍射峰峰强明显增强,且在18.7°处出现了 ZrO2衍射峰,说明 ZrO2的引入促进了CeO2和Ce2O3间的相互作用.如图6(c),不同焙烧温度下(300、400、500℃),随着焙烧温度升高,堇青石在54.9°处的衍射峰发生了向左偏移,从 54.9°偏移到54.1°,说明催化剂在温度升高后其催化剂结构发生了变化,可能与催化剂孔道结构发生显著变化有关.

图6 不同条件制备的催化剂的XRD图Fig.6 XRD patterns of catalysts prepared under different conditions
如表 1和图 7所示,对于同一种催化剂载体Al2O3,不同铬铈比例催化剂显示出不同的表面结构特性,当Cr/Ce物质的量比为1:2时,比表面积及孔径均呈现出最大值,展现出较好的氧化活性;当催化剂掺杂 ZrO2后其比表面积进一步增大,而孔体积和平均孔径减少,表明掺杂 ZrO2有利于催化剂氧化性能的提高.相较于Al2O3和ZSM-5载体,以堇青石为载体的Cr2O3-CeO2-ZrO2比表面积、孔体积和平均孔径均显现出最大值,这与催化剂活性测试结果相一致.由图7可知,载体为堇青石的催化剂具有II型气体等温吸附线,且相对压力P/P0在0.7~1.0范围内具有 H3型滞后环,高压区吸附量上升较快,说明孔径尺寸较大、分布相对较宽且大孔居多[22],有利于催化剂活性氧化物吸附反应物以及反应产物的排出,而对载体为 ZSM-5和Al2O3的催化剂而言,孔径分布较窄,在4~6nm之间.

图7 部分样品的吸附等温线及孔径分布Fig.7 Adsorption isotherms and pore size distribution of some samples

表1 部分样品比表面积及孔容孔径Table 1 Specific surface area, pore volume and pore diameter of samples
由图8可知,以Al2O3为载体(图8(B))的催化剂表面出现大量针状或片状活性组分,较为分散,而以堇青石(图8(A))与ZSM-5(图8(C))为载体的催化剂活性组分以球状颗粒物形式存在,且与载体结合较为紧密,表明载体对催化剂活性组分形态结构产生重要影响.对于 Cr2O3-CeO2-ZrO2(Zr/Cr=0.5)/堇青石催化剂,随着焙烧温度由 300℃(图 8(D))升高至400℃(图 8(E)),其表面所负载的活性组分物质的量显著增加且分布更加均匀,而当焙烧温度进一步升高至 500℃(图 8(F))时,活性组分因高温导致熔融现象的发生,孔径增大,比表面积减小,致使催化剂氧化活性降低.

图8 催化剂的SEM图Fig.8 SEM of catalyst
2.2.2 H2-TPR 如图 9所示,对于 Cr2O3-CeO2/Al2O3,掺杂有NiO和ZrO2的催化剂在261,265℃显现出低温 H2还原峰,其还原峰可能分别归属为催化剂中 Ni2+还原峰和 Cr3+,Zr4+组合还原峰,而 482℃处显示的是单组分Cr2+的H2还原峰[23],且掺杂ZrO2的催化剂展现出更大的 H2还原特征峰面积,促使 Cr3+的H2还原峰向低温转移而形成组合还原峰,Cr2O3和ZrO2间的相互作用促进了催化剂氧化还原能力提升,有利于催化剂氧化活性的发挥.而对于掺杂有 CuO,FeOx,CoOx和 FeOx催化剂而言,所掺杂的氧化物 H2还原峰位于 290~305℃间,而 Cr2O3则显示出更高的H2还原峰,且 CeO2的 H2还原峰(>788℃)更高,掺杂的过渡金属氧化物未能与Cr2O3或CeO2间产生相互作用,均以混合氧化物的形式存在,不利于催化剂氧化性能的提高.对于Cr2O3- CeO2/堇青石,261℃和265℃处所呈现的H2还原峰分别为Cr3+还原为Cr2+和Cr2+还原为 Cr0,而对于 Cr2O3-CeO2-ZrO2/堇青石催化剂在278,357和490℃所展示的H2还原峰为Cr3+和Zr4+组合还原峰,且H2还原峰温度明显低于Cr2O3-CeO2/堇青石催化剂,表明Cr2O3和ZrO2间存在强烈的相互作用而有助于提升催化氧化性能,与 Cr2O3-CeO2-ZrO2/Al2O3催化剂相一致.

图9 过渡金属氧化物掺杂的Cr2O3-CeO2/Al2O3和Cr2O3-CeO2/堇青石催化剂H2-TPR图Fig.9 H2-TPR diagram of transition metal oxide doped Cr2O3-CeO2/Al2O3 and Cr2O3-CeO2/cordierite catalysts
2.2.3 XPS 图 10(a)中,位于 904.1,886.2eV 处的谱峰分别属于Ce3+的3d5/2和3d3/2,其余谱峰均为Ce4+的特征峰.随着 ZrO2掺杂量的增大,Ce3+的特征峰向低结合能偏移,表明ZrO2掺入可能引起Ce2O3粒子半径的增大.图10(b)中,均显示出3个矮峰,随着ZrO2的掺杂量增大,Cr4+2p3/2结合能由 580.1eV迁移至579.3eV,这是由于大量 ZrO2的掺杂改变了催化剂结构中的电荷平衡,其中 Ce3+/Ce4+随着 Zr掺量的增加分别为 0.199、0.208和 0.305,说明部分 Cr3+与 Ce4+在焙烧过程中发生电子交换,致使Ce3+的含量增加[24].由图10(c)可知,Cr2O3-CeO2-ZrO2(Zr/Cr=0.5)/堇青石催化剂具有更多的表面活性氧(Osur),占 39.9%,其氧化活性亦最佳,显然这是由于其表面 Ce3+存在,使得大量的氧空位形成以维持催化剂表面电中性平衡所致[25-26].在氯苯以及大部分 VOCs催化氧化反应中,具有更多氧空位的催化剂,因表面活性吸附氧的增加,往往具有优异的氧化性能.Cr2O3-CeO2-ZrO2(Zr/Cr=0.75)/堇青石催化剂具有更多的表面羟基氧(Oads),其中Oads/Olat随着Zr掺量的增加分别为1.54、1.67和2.85,说明晶格氧(Olat)含量随着ZrO2掺量的增加而减少,这可能是由于过量 ZrO2的掺入导致部分晶格氧存在于体相中.图10(d)中184.7,182.2eV处的谱峰分别属于 Zr 3d3/2和 Zr 3d5/2,随着 ZrO2掺杂量的增大,Zr4+的状态未发生显著变化.
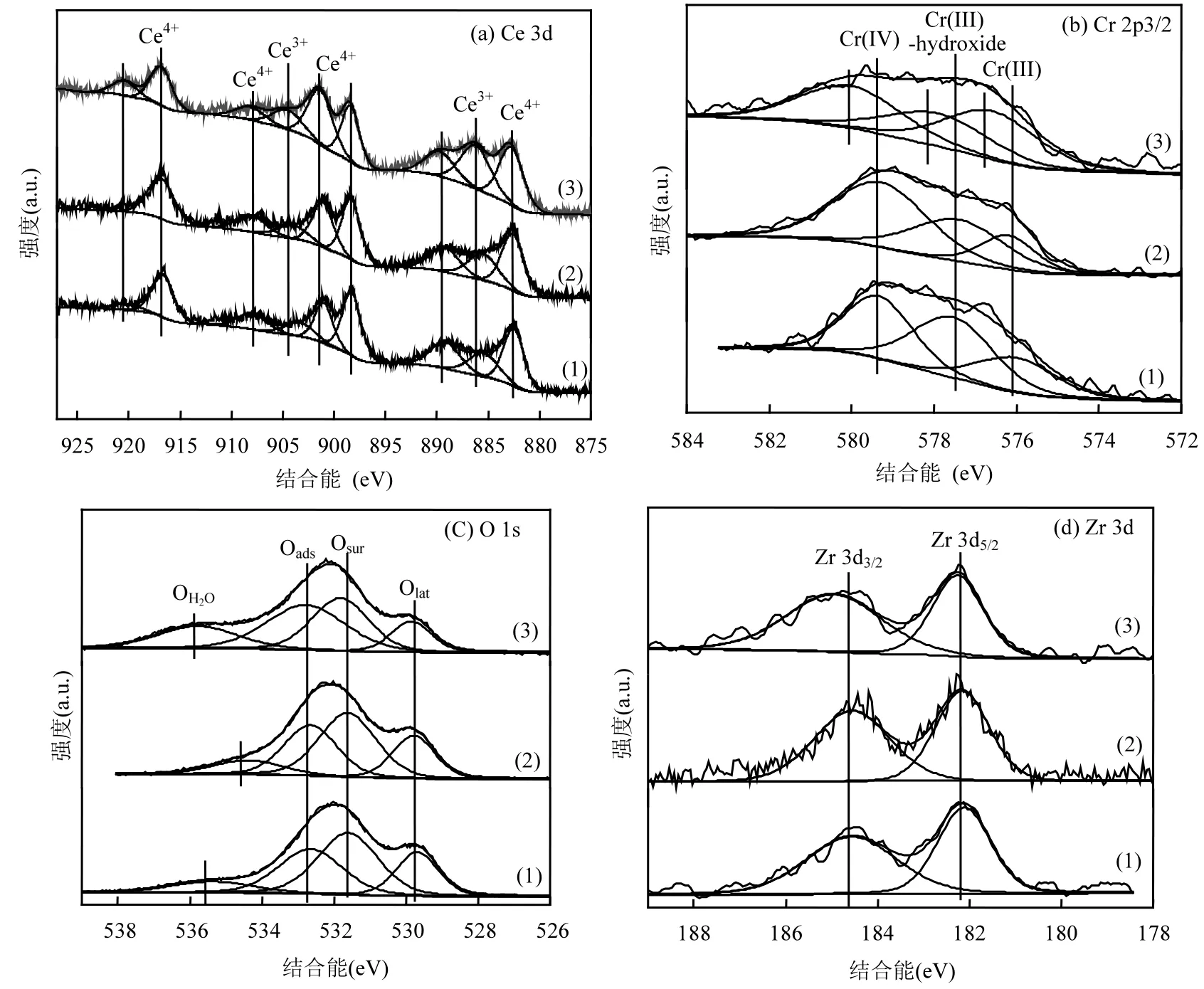
图10 Cr2O3-CeO2-ZrO2/堇青石催化剂XPS图Fig.10 XPS of Cr2O3-CeO2-ZrO2/cordierite catalyst
3 结论
3.1 催化剂载体对催化剂活性组分形态结构产生重要影响.以堇青石为载体的 Cr2O3-CeO2-ZrO2催化剂表现最佳,T90=350℃,且24h稳定性测试表明催化剂催化氧化氯苯转化率始终维持在 99.0%以上,无明显氯中毒现象的发生,其主要是由于Cr/Ce物质的量比为 1:2时,氧化铬和二氧化铈间形成了CrCeOx固溶体.
3.2 掺杂有ZrO2的催化剂展现出更低的还原温度和更大的H2还原峰面积进而形成组合还原峰,Cr2O3和 ZrO2间的相互作用促进了氧化还原能力提升.且随着 ZrO2掺杂量的增大,Ce3+的特征峰向低结合能偏移,这是由于大量 ZrO2的掺杂改变了催化剂结构中的电荷平衡,部分 Cr3+与 Ce4+在焙烧过程中发生电子交换,致使 Cr4+及 Ce3+的含量增加,氧空位增加促使表面活性氧(Osur)增多,有利于催化剂催化氧化性能的提高.
