原料乳中产蛋白酶假单胞菌双重PCR检测体系建立和评价
2021-08-02吴天赐李楠张娟余意刘振民
吴天赐,李楠,张娟,余意,刘振民*
1(光明乳业股份有限公司乳业生物技术国家重点实验室,上海乳业生物工程技术中心,上海,200436)2(上海大学 生命科学学院,上海,200444)
假单胞菌是原料乳中普遍存在的嗜冷菌菌群,因其能在低温条件下正常生长繁殖,产生耐热蛋白酶和耐热脂肪酶,被认为是原料乳中具有极大危害性的菌群之一[1-3]。假单胞菌所产的碱性金属蛋白酶(alkaline metalloproteinase)是其中一种常见的耐热胞外蛋白酶,这种酶受到aprX基因的调控且无法通过巴氏杀菌和超高温灭菌完全灭活,可导致乳制品在货架期内出现凝乳、发苦等[4]。随着研究不断深入,发现多类假单胞菌中存在相同或类似的蛋白酶基因,这些假单胞菌对原料乳及乳制品存在一定的潜在危害[5-7]。
我国对于原料乳中嗜冷假单胞菌的检测方法依赖于传统的平板计数方法[8],主要根据理化性质来判断,此方法存在检测周期长、重复性不好和容易漏检等缺点,很容易错过检测的最佳时期。PCR检测技术因其快速、高效等特点已经被运用于食品中危害性微生物的检测。将16S rRNA[9]和rpob基因[10]等作为特异性靶点进行PCR检测假单胞菌已被广泛报道。sucD基因是琥珀酰辅酶A合成酶的α亚基编码基因[11],以sucD基因作为靶点检测假单胞菌的研究非常少。研究[12]发现sucD基因在假单胞菌属内存在较高的同源性,可作为假单胞菌检测靶点。此外,MARCHAND等[13]研究表明将aprX基因作为靶点能够稳定可靠的检测假单胞菌。
多重PCR技术是PCR技术的一种,在一个反应体系中加入2对或2对以上引物,可同时针对多个靶点进行扩增[14]。因为多重PCR与常规单一PCR相比,具有省时、低成本和高特异性等优点,在临床医学、农业和食品行业等领域的检测都已得到广泛的运用[15-17]。随着我国乳制品行业引入冷链系统后,假单胞菌类嗜冷菌对原料乳的危害越发明显,但迟迟没有合适的解决方案。本研究将致力于针对2个目的基因,建立一种能够快速检测原料乳中具有产蛋白酶能力假单胞菌的双重PCR检测方法,并对反应体系进行优化和评价。
1 材料与方法
1.1 材料、试剂和仪器
实验所用菌株见表1,菌株均为-80 ℃甘油保存。

表1 实验菌株Table 1 Strains used in the study
细菌基因组DNA提取试剂盒、通用型DNA纯化回收试剂盒,天根生化科技有限公司;100 bp DNA Marker以及TaqDNA聚合酶等PCR反应试剂,Takara;酵母提取物,阿法埃莎(Alfa Aesar)化学有限公司;牛肉膏,上海盛思生化科技有限公司;蛋白胨,国药集团化学试剂有限公司;NaCl固体、磷酸二氢钾、磷酸二氢钠(均为分析纯)、蛋白肽,国药集团化学试剂有点公司;特异性引物合成及基因测序由生工生物工程(上海)有限公司完成。
NB培养基:0.3%(质量分数,下同)牛肉膏,1%蛋白胨,0.5% NaCl固体,2%琼脂粉,pH为7.0;脱脂乳培养基:0.5%酵母提取物、2%脱脂乳粉、1.5%琼脂粉;增菌液[18]:1%蛋白胨,0.5% NaCl固体,0.35%磷酸二氢钠,0.15%磷酸二氢钾,最终pH为7.2±0.2。
Veriti Thermal Cycler 96-well PCR仪,美国Applied Biosystems公司;BIO-RAD GelDoc XR+凝胶成像系统,上海安景科技有限公司;HVE-50型高压灭菌锅,日本HIRAYAMA公司;SG-402TX型超净工作台,美国THE BAKER COMPA-NY公司;Minispin Plus离心机,德国Eppendorf AG公司;HE-120核酸电泳仪,上海天能科技有限公司;AL104电子天平,梅特勒-托利多仪器(上海)有限公司;PHS-25型pH计,美国奥立龙公司;XW-80A型漩涡混合仪,上海青浦沪西仪器厂;HZQ-X500C恒温振荡培养箱,上海一恒科学仪器有限公司;NanoDrop 2000C分光光度计,美国Thermo Fisher Scientific公司。
1.2 实验方法
1.2.1 引物设计
根据文献中提到的蛋白酶基因aprX和琥珀酰辅酶A合成酶α亚基编码基因SucD,在NCBI数据库中下载多条假单胞菌株靶点基因序列,利用MEGA X软件进行ClustalW序列对比,分析基因的保守序列,再应用Primer premier 5.0和Oligo 7对保守序列进行引物设计,尽量减少引物间的二聚体形成,并分析了最佳退火温度,通过NCBI中Primer-BLAST功能对比,筛选了2对特异性较好的引物(表2)。引物由上海生工生物工程有限公司合成,扩增片段大小依次为232、415 bp。
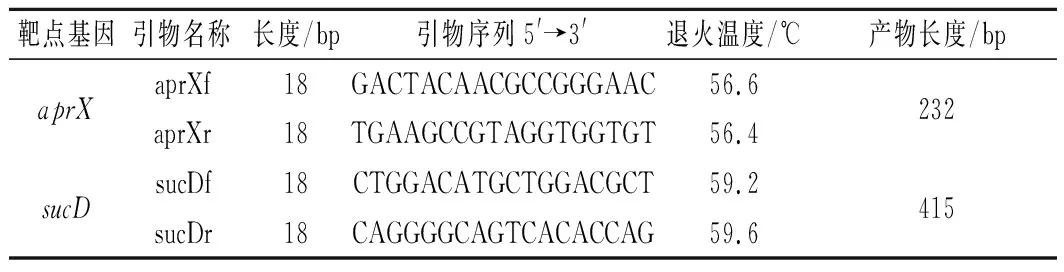
表2 PCR引物信息Table 2 Information about the PCR primers
1.2.2 细菌基因组DNA提取
不同的实验中,分别采用试剂盒法或煮沸法[9]提取细菌基因组DNA。
测试菌株采用试剂盒法提取基因组DNA,使用推荐固体培养基和培养温度活化,挑取单菌落接种至液体培养基在恒温振荡培养箱培养24 h,取1.5 mL菌液,按照天根生化科技有限公司的细菌基因组DNA提取试剂盒操作说明进行细菌基因组DNA提取,保存于-20 ℃下,作为PCR模板。原料乳及模拟样品采用煮沸法提取基因组DNA:取8 000×g离心5 min,弃上清液,用200 mL无菌水重悬菌体,在100 ℃煮沸10 min,立即放入-20 ℃冰箱冷却,20 000×g离心10 min,上清液即为细菌基因组DNA。
1.2.3 单重PCR反应体系建立
单重PCR反应体系(50 μL):10×Buffer 5 μL,dNTP(deoxy-ribonucleoside triphosphate) Mixture(各2.5 mmol/L)4 μL,Taq酶(5 U/μL)0.25 μL,上下游引物(10 μmol/L)各1 μL,DNA模板1 μL(以产氮假单胞菌BNCC 133928基因组DNA为模板,此类为前期从原料乳中分离嗜冷菌时出现较多的菌),ddH2O补足50 μL。反应条件如下:94 ℃预变性5 min;94 ℃变性30 s,58 ℃退火30 s,72 ℃延伸1 min,循环30次;72 ℃延伸10 min。PCR扩增产物,采用1%(质量分数)琼脂糖凝胶(含Gelred染料),在100 V下电泳30 min,将目的条带割胶回收送至上海生工测序。
1.2.4 双重PCR反应体系建立及条件优化
以标准菌株BNCC 133928产氮假单胞菌基因组DNA作为模板,与各种PCR反应试剂混合,主要针对退火温度、引物浓度配比和Taq酶量来完成双重PCR反应体系建立和条件优化。
1.2.5 特异性检测
将表1中所有菌株的基因组DNA作为模板,进行双重PCR扩增,并以ddH2O作为空白对照。以此判断此方法对常见致病菌和原料乳中常见细菌的特异性。同时阳性条带割胶回收送至上海生工测序。
1.2.6 灵敏度检测
使用NanoDrop 2000超微量分光光度计测定标准菌株产氮假单胞菌DNA核酸浓度,使用ddH2O进行10倍梯度稀释,分别取不同浓度DNA 各1 μL作为模板进行双重PCR,检测双重PCR反应体系的灵敏度。
1.2.7 人工模拟污染原料乳检测
将活化的产氮假单胞菌接种1环至NB培养基中30 ℃下培养24 h,10倍梯度稀释,对10-5、10-6、10-7、10-8、10-9稀释梯度进行平板计数,平行3次。参照MARTINS等[19]的方法模拟原料乳污染实验,将菌液用120 g/L脱脂乳梯度稀释,得到菌液浓度为101、102、103、104、105和106CFU/mL的模拟原料乳样品,各取1 mL 利用煮沸法。将所提DNA分别进行双重PCR检测,以ddH2O作为阴性对照。模拟工业检测中增菌处理,在225 mL增菌液中加入25 mL已灭菌的120 g/L脱脂乳,分别接入1 mL浓度为100、101、102和103CFU/mL菌液,平行3次,在30 ℃,150 r/min条件下培养,分别在0、4、8、12和24 h各取1 mL样品,用煮沸法提取细菌基因组DNA,进行双重PCR检测。
1.2.8 实际原料乳样品检测
共15份来自北京、上海、广州牧场的原料乳样品,每份原料乳样品取25 mL加入225 mL增菌液中,在30 ℃、150 r/min条件下摇床增菌培养24 h。采用煮沸法提取DNA,分别作为模板,利用双重PCR方法进行检测。同时采用培养方法在6.5 ℃下培养7 d筛选可疑菌落,平行3次,以防漏检,对筛选的菌株进行16S rDNA 测序鉴定,并用20 g/L脱脂乳培养基验证筛选的假单胞菌是否具有产蛋白酶能力作为对照试验。
2 结果与分析
2.1 单重PCR特异性检测结果
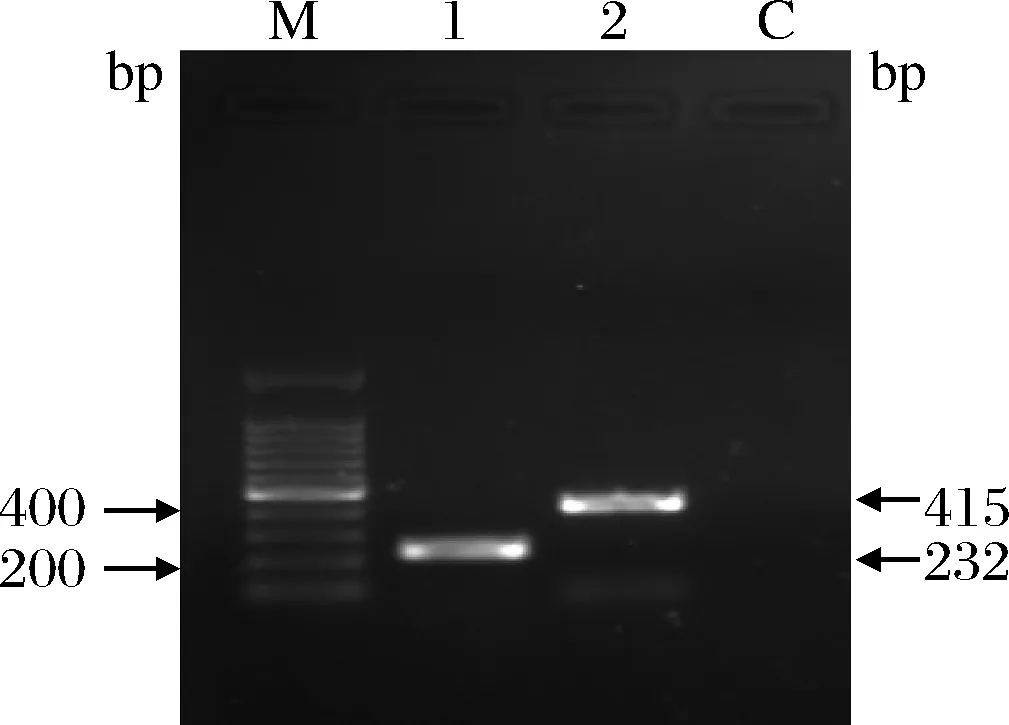
使用单重PCR反应验证引物的特异性,aprX引物aprXf/aprXr的特异性验证,结果仅在232 bp处扩增出了预期的产物,sucD引物sucDf/sucDr的特异性验证,结果仅在415 bp处扩增出了预期的产物,如图1所示。上述阳性条带割胶回收送至上海生工生物工程有限公司测序,测序结果在NCBI中BLAST在线对比,证实扩增条带为目的片段。
2.2 双重PCR反应体系建立及优化
通过对退火温度、Taq酶量和引物浓度配比优化后,得到最佳退火温度为57 ℃、最佳Taq酶量为0.5 μL、最优引物浓度配比为aprX10 μmol/L,sucD7.5 μmol/L,如图2所示。最终双重PCR反应体系为:10×Buffer 5 μL,dNTP Mixture(各2.5 mmol/L)4 μL,Taq酶(5 U/μL)0.5 μL,aprXf/aprXr各1 μL(10 μmol/L),sucDf/sucDr各1 μL(7.5 μmol/L),DNA模板1 μL,ddH2O补足50 μL。反应条件为:94 ℃ 5 min;94 ℃ 30 s,57 ℃ 30 s,72 ℃ 1 min,30个循环;72 ℃ 10 min。

2.3 双重PCR特异性检测结果
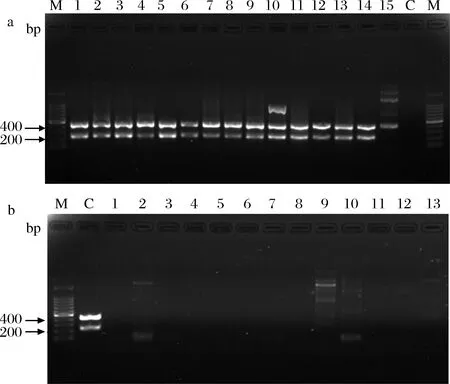
表1中所有菌株基因组DNA进行双重PCR反应,结果显示在目标区域内1~14号阳性菌株均能扩增出2条清晰的目的条带;15号恶臭假单胞菌在目标区域内扩增出1条415 bp条带(30 ℃下培养48 h不产蛋白酶,图3);其余阴性菌株均未扩增出目的片段,如图4所示,经测序结果在线比对,证实确为目的片段,表明本实验双重PCR方法特异性良好。

2.4 灵敏度检测结果
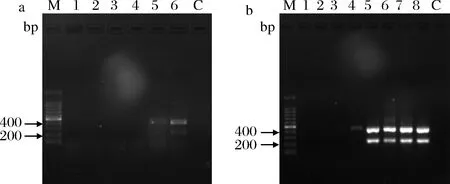
所提取的标准菌株产氮假单胞菌基因组DNA以10倍梯度稀释至10-6,以各梯度为DNA模板进行双重PCR反应,结果如图5所示,当反应体系中DNA模板最终质量浓度为1.67×10-2pg/μL时,未能扩增出目标条带,因此本方法的检测限为1.67×10-1pg/μL。

2.5 人工模拟污染原料乳样品的检测结果
培养24 h后所得到的菌液初始浓度为3.5×108CFU/mL,将菌液用120 g/L脱脂乳分别稀释至3.5×101、3.5×102、3.5×103、3.5×104、3.5×105和3.5×106CFU/mL,模拟被假单胞菌污染的原料乳,各取1 mL采用煮沸法提取细菌基因组DNA后进行双重PCR反应,结果当菌液浓度达到3.5×106CFU/mL时,能够清晰检测出2条目的条带。当样品经过增菌后,发现当增菌12 h后,在菌液浓度为3.5×103CFU/mL 的样品中检测到了2条阳性条带,当增菌24 h后,在所有样品中均检测出了2条阳性条带,如图6。所以在适当的增菌处理后,本方法的检测限可达到3.5 CFU/mL,检测灵敏度高且效果良好。
2.6 实际原料乳样品检测
采集15份原料乳样品,检测结果如表3所示。经培养法筛选后,测序检测及验证,结果仅有5份样品检出产蛋白酶假单胞菌,检出率为30%。而通过双重PCR方法,有10份样品检出2条清晰阳性条带且无非特异性扩增,检出率为60%。相比之下,本研究所建立的方法更加省时且准确率高。

表3 原料乳样品检测结果Table 3 Detection results of the raw milk samples
3 结论与讨论
目前我国乳制品行业的冷链运输技术没有达到成熟阶段,无法保证所有环节的冷藏温度不产生较大的波动,这可能导致假单胞菌的大量繁殖并产生蛋白酶[20]。原料乳中危害性假单胞菌在运输贮藏等过程中所产生的耐热蛋白酶等无法通过巴氏灭菌和超高温灭菌完全清除,这是产蛋白酶假单胞菌对乳造成危害的主要原因之一[21]。快速检测原料乳中具有危害性的假单胞菌并通过一定手段进行控制,将有效地提高原料乳品质。在实际工业检测中,与传统的PCR检测技术相比,双重PCR方法能够有效避免“假阳性”,并且具有省时、特异性高、灵敏性高等特点。
本研究结合NCBI数据库中多种假单胞菌的sucD基因和aprX基因序列,利用软件进行序列分析,筛选出2对具有特异性较高且适用于双重PCR的引物,建立了检测效果显著的双重PCR方法。胡冰雪等[22]针对荧光假单胞菌gyrB基因、沙门氏菌invA基因和单增李斯特菌的hlyA基因所建立的m-PCR,经优化后纯菌DNA检测限为1 pg/μL,增菌培养后荧光假单胞菌、沙门氏菌和单增李斯特菌检测水平分别为9、5和70 CFU/mL。本研究建立的方法对纯菌DNA的检测限为0.167 pg/μL,经24 h增菌培养后检测限可达到3.5 CFU/mL。
本方法经过验证具有很高的特异性和灵敏度,相对于传统培养筛选的方法能够节省大量时间、试剂等。实际检测中无需使用试剂盒,仅通过煮沸法提取DNA即可进行检测,可有效的降低成本,能够满足工业中快速检测的要求。此方法可应用于乳制品行业的原料乳中产蛋白酶假单胞菌检测,为奶业等控制产蛋白酶假单胞菌具有借鉴价值。
