调控质粒拷贝数优化酿酒酵母异源合成真菌聚酮10,11-dehydrocurvularin
2021-08-02严豪王志远庞子萱林苹鑫吴疆李业白仲虎
严豪,王志远,庞子萱,林苹鑫,吴疆,李业*,白仲虎*
1(江南大学 生物工程学院,江苏 无锡,214122)2(粮食发酵工艺与技术国家工程实验室(江南大学),江苏 无锡,214122)
聚酮类化合物(polyketides)是细菌、真菌及植物产生的一类次级代谢产物[1],由聚酮合酶(polyketide synthase,PKSs)以酰基-辅酶A(coenzyme A,CoA)为前体进行连续缩合,经环化、修饰最后释放的一类天然产物。其中真菌是该类次级代谢产物的主要来源[2]。许多聚酮类化合物因其具有重要而丰富的生物活性而在临床上得到了广泛的应用,如抗菌的红霉素、抗癌药物阿霉素、杀虫剂多杀菌素、抗寄生虫药物阿维菌素以及免疫抑制剂雷帕霉素等[3]。10,11-dehydrocurvularin属于二羟基苯乙酸内酯[4](dihydroxyphenylacetic acid lactones,DAL)类化合物,其因具有热休克活性拥有优异的广谱抗肿瘤活性[5]。目前的研究方向大多为改造聚酮合酶以获得结构多样性的活性分子[6-7],已发现的一些聚酮类活性化合物普遍存在生产成本高、天然宿主产量低的问题。
近年来,微生物代谢工程的发展使得一些生产成本昂贵的药物低成本化、高效率化[8]。酿酒酵母作为最简单且安全的真核模式菌株[9],人们对其的分子生物学改造手段及应用十分丰富。酿酒酵母在食品和发酵过程中用于生产酒精饮料和面包的历史悠久[10],已被广泛用于代谢途径改造、外源合成真核生物代谢产物,是食品、医药和生物能源方面的重要生产用微生物[11]。酿酒酵母BJ5464-NpgA(MATαura3-52his3-Δ200leu2-Δ1trp1pep4∶HIS3prb1Δ1.6Rcan1GAL)是一种生产聚酮类化合物的合适宿主[12]。Pep4和Prb1的功能缺陷能有效降低外源蛋白的降解;引入了一个来源于其他真菌中的泛酰基转移酶NpgA,该酶能够活化真菌聚酮类化合物合成酶(PKSs)里的酰基载体蛋白(acyl carrier protein,ACP)结构域。
质粒是游离在基因组之外的环状DNA分子,质粒的拷贝数越高,意味着跟随质粒的外源基因复制越多,相对而言外源基因表达得到的蛋白就会越多。同时,通过可控质粒拷贝数,调整多酶体系的不同酶的通量,优化目标产物的代谢途径。对于质粒拷贝数的研究,目前,LIAN等[13]以酿酒酵母INVSc1为出发菌株,筛选出了具有拷贝梯度的几种筛选标记,使用该体系组合优化正丁醇代谢途径,产量较出发菌株提高了100倍。也有研究构建了拷贝数依赖的方式在酵母中表达赤点石斑鱼坏死病毒衣壳蛋白,与野生型菌株相比产量增加了150倍[14]。本研究在酿酒酵母BJ5464-NpgA异源表达来源于Aspergillusterreus的聚酮化合物合酶(S1与S2),以得到产物10,11-dehydrocurvularin,并通过截短启动子构建转录结合缺陷的整合盒,使用抗生素筛选压调控质粒拷贝数,以此优化产物合成途径获得更高产量。
1 材料与方法
1.1 菌株、质粒和引物
酿酒酵母BJ5464-NpgA、原始质粒pXK30-FLAG 30F、pXW06-FLAG 06F,携带土曲霉来源的聚酮合酶基因片段的质粒pYX003-pXW06F_AtF7_RedPKS、pYX004-pXK30F_AtF7_nrPKS来自亚利桑那大学;质粒pYTK-034、pYTK-077、pYTK-079通过Addgene向加州大学伯克利分校生物工程系购买,所用的大肠杆菌DH5α由本实验室保藏。在本研究中使用的质粒如表1所示,本文中所用到的引物见表2,因KanMX和HygB抗生素表达盒的启动子均为AgTEF Promoter,其20 bp截短及更长的截短的扩增引物均完全在启动子上,两者的5′到3′端引物可通用;且两者所用终止子均为AgTEF Terminator,其截短标记的3′到5′端引物可通用。引物合成以及质粒测序由苏州金唯智生物科技有限公司完成。

表1 本研究使用的质粒Table 1 Plasmids used in this study

表2 本研究所用的引物Table 2 Primers in this study
1.2 酶和试剂
所有限制性内切酶,Thermo Fisher Scientific;2×Es Taq MasterMix(Dye),北京康为世纪生物科技有限公司;DNA高效连接液,TaKaRa公司;同源重组酶、HiScript II Q Select RT SuperMix for qPCR,诺唯赞公司;质粒提取试剂盒、PCR产物纯化试剂盒、胶回收试剂盒,Axygen公司;遗传霉素、潮霉素、酵母DNA提取试剂[V(酚)∶V(氯仿)∶V(异戊醇)=25∶24∶1],北京索莱宝科技有限公司;氨苄青霉素,上海生工;酵母-Trp/-Ura双缺培养基,上海懋康生物科技有限公司;10,11-dehydrocurvularin标准品,Toronto Research Chemicals。
1.3 培养基和摇瓶培养条件
大肠杆菌使用LB培养基在37 ℃培养,抗生素使用氨苄青霉素,终质量浓度为50 μg/mL。酿酒酵母使用YPD(质量分数为1%酵母提取物+2%蛋白胨+2%葡萄糖)与合成培养基在250 mL 摇瓶中培养,装液量50 mL,接种量2%,培养温度为30 ℃,转速为220 r/min。使用深孔培养板进行培养时,装液量为2 mL,其他条件同上。
1.4 质粒构建
为得到携带mruby2荧光的表征载体pBL001-30F-mruby2,用引物mruby2-F/mruby2-R扩增两端带有pXK30-FLAG 30F质粒的同源臂的mruby2基因片段。将扩增片段与SwaI内切酶线性化过的pXK30-FLAG 30F质粒利用同源重组酶重组,转化至大肠杆菌感受态,挑取单克隆测序验证。
用内切酶PstI和XbaI线性化质粒pBL001-30F-mruby2。利用相应引物对质粒pYTK-077扩增得到不同截短长度启动子的KanMX表达盒片段,对质粒pYTK-079扩增得到不同截短长度启动子的HygB表达盒片段,将表达盒片段使用内切酶PstI和XbaI酶切处理后,使用DNA连接酶连接线性化质粒pBL001-30F-mruby2和表达盒片段,转化大肠杆菌,测序验证得到30F-mruby2-Kan系列和30F-mruby2-Hyg系列质粒。
用内切酶SbfI对pYX003-pXW06F_AtF7_RedPKS和pYX004-pXK30F_AtF7_nrPKS进行线性化。利用相应引物对质粒pYTK-077扩增得到不同截短长度启动子的KanMX表达盒片段,对质粒pYTK-079扩增得到不同截短长度启动子的HygB表达盒片段,将表达盒片段使用内切酶SbfI酶切处理后,使用DNA连接酶连接线性化质粒pYX003-pXW06F_AtF7_RedPKS和不同截短长度启动子的KanMX表达盒片段,不同截短长度启动子的HygB表达盒片段与线性化质粒pYX004-pXK30F_AtF7_nrPKS连接,转化至大肠杆菌感受态,挑取单克隆测序验证得到06F-003-Kan系列质粒和30F-004-Hyg系列质粒。
1.5 mruby2荧光蛋白强度的测定
将质粒pBL001-30F-mruby2、pXW06-FLAG 06F使用醋酸锂转化法[16]转化至BJ5464-NpgA,使用-Trp/-Ura双缺缺陷型固体培养基进行筛选,每个转化从琼脂培养基上挑取3个单克隆,使用24孔深孔板在合成培养基中进行预培养,以2%转接至含300 mg/L相应抗生素的YPD培养基中培养24 h,吸取10 μL发酵液加190 μL去离子水稀释在酶标板里,使用酶标仪测定OD600和荧光强度。激发波长为559 nm,发射波长为600 nm。
1.6 总DNA的提取及实时荧光定量PCR
收集约OD600=20的菌体至破碎管中,加入与细胞体积相当的酸性玻璃球,加400 μL STES(20 mmol/L Tris,50 mmol/L NaCl,1 mmol/L EDTA,pH=7.5)溶液,再加400 μL 酵母DNA提取试剂,剧烈振荡10×30 s,每次间隔30 s,置于冰上。加入200 μL TE吹吸混匀,10 000 r/min离心5 min,溶液分4层,吸取最上层300 μL至1.5 mL离心管中,加入10%体积分数的3.0 mol/L醋酸钠,2倍体积无水乙醇,稍混匀,在-80 ℃静置30 min后,10 000 r/min离心10 min弃上清液。加入500 μL无水乙醇洗涤,10 000 r/min离心5 min,弃尽上清液,55 ℃干燥DNA 20 min,加入40 μL TE(含RNase 20 μg/mL),在55 ℃水浴30 min以消化RNA,得到无RNA杂质的酿酒酵母总DNA。
得到上述的总DNA后,将其质量浓度稀释为100 ng/μL,用于qPCR分析。以染色体上的ALG9基因作为内参基因,使用绝对定量法[17-18]分别测定目标基因KanamycinR和HygromycinR的拷贝数与Ct值的标准曲线。
1.7 在酿酒酵母BJ5464-NpgA中合成10,11-dehydrocurvularin
将携带Aspergillusterreus来源的聚酮合酶基因片段的质粒pYX003-pXW06F_AtF7_RedPKS、pYX004-pXK30F_AtF7_nrPKS转化至BJ5464-NpgA[15],每个转化从琼脂培养基上挑取3个单克隆,在多孔板接种2 mL合成培养基中,30 ℃,220 r/min 摇床培养1 d。随后以2%的接种量转接至2 mL合成培养基中,30 ℃,220 r/min 摇床培养2 d。
1.8 发酵产物的处理及HPLC检测方法
使用盐酸将发酵液pH值调节至5,在12 000 r/min 下离心10 min,用等体积的乙酸乙酯萃取上清液3次,取500 μL进行真空离心浓缩,浓缩物使用100 μL甲醇溶解,12 000 r/min离心10 min,取上清液进行HPLC分析,进样量为20 μL。
HPLC分析程序:0~5 min流动相为体积分数为5%的乙腈和95%的0.1%乙酸溶液,5~15 min流动相为乙腈在0.1%乙酸溶液的线性梯度为5%~95%,15~25 min流动相为95%的乙腈和5%的0.1%乙酸溶液;流速为0.8 mL/min;岛津inertsustain®C18色谱柱(5 μm,250 mm×4.6 mm);检测波长为300 nm[19]。
1.9 优化10,11-dehydrocurvularin合成途径以提高产量
将携带Aspergillusterreus来源的聚酮合酶基因片段且整合了抗生素抗性基因整合盒的质粒转化至BJ5464-NpgA,每个转化从琼脂培养基上挑取3个单克隆,使用24孔深孔板在SC培养基中进行预配养,以2%接种量转接至添加相应抗生素的SC培养基中培养48 h,发酵液的处理与产物检测方法如上。
2 结果与分析
2.1 转录结合缺陷抗生素抗性基因整合盒的构建
将mruby2基因利用同源重组整合到pXK30-FLAG 30F得到质粒pBL001-30F-mruby2,经测序验证后,在质粒pBL001-30F-mruby2基础上构建转录结合缺陷抗生素抗性基因整合盒如图1-a和图1-b所示,将构建的质粒分别用XbaI和PstI酶切,结果如图1-c和图1-d所示,所的条带大小与理论值相符,测序结果表明表达盒已成功构建。

2.2 转录结合缺陷抗生素抗性基因整合盒的mruby2表征
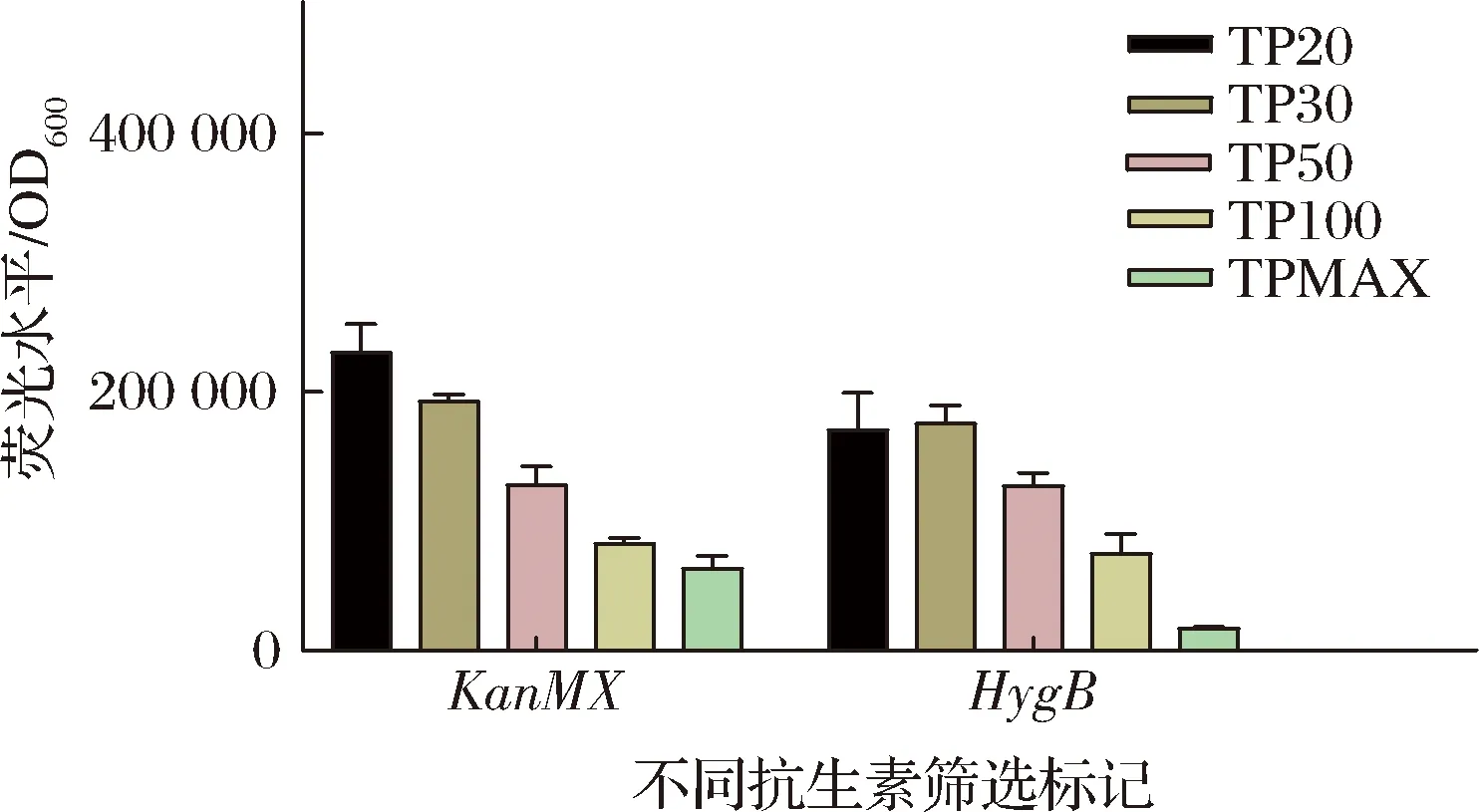
将带有mruby2的整合盒里的质粒转化至BJ5464-NpgA表达,在抗生素质量浓度都为300 mg/L的YPD中发酵,结果如图2所示。结果表明,截短抗生素抗性基因的启动子会使得在同一个质粒上的荧光蛋白基因表达增强,且荧光蛋白的强度与启动子长度一定程度上呈负相关。
2.3 拷贝数绝对定量的标准曲线的建立
根据已报道的方法分别构建2种基因标准曲线所需的单拷贝质粒。在KanamycinR基因和ALG9基因是单拷贝的质粒中,KanamycinR基因和ALG9基因标准曲线见图3-a,KanamycinR基因标准曲线的回归方程为y=-3.221x+35.92(R2=0.996 3),ALG9基因的标准曲线的回归方程为y=-3.329x+35.83(R2=0.995 9);在HygromycinR基因和ALG9基因是单拷贝的质粒中,HygromycinR基因和ALG9基因的标准曲线见图3-b,HygromycinR基因标准曲线的回归方程为y=-3.267x+36.75(R2=0.996 0),ALG9基因的标准曲线的回归方程为y=-3.344x+36.28(R2=0.994 2)。
2.4 实时荧光定量PCR测定质粒拷贝数
按照方法1.6提取带荧光蛋白的重组菌的总DNA并进行实时荧光定量PCR测定质粒拷贝数,经定量换算后的质粒拷贝数结果如表3和表4所示。从结果中可以看出,通过截短抗生素抗性基因的启动子,转录结合缺陷使得其耐抗生素能力下降,细胞会通过增加质粒拷贝数来维持一定水平的耐抗生素能力。在一定范围内,荧光强度随着抗生素抗性基因启动子越短而增强,是因为荧光蛋白基因mruby2的拷贝数在增加。
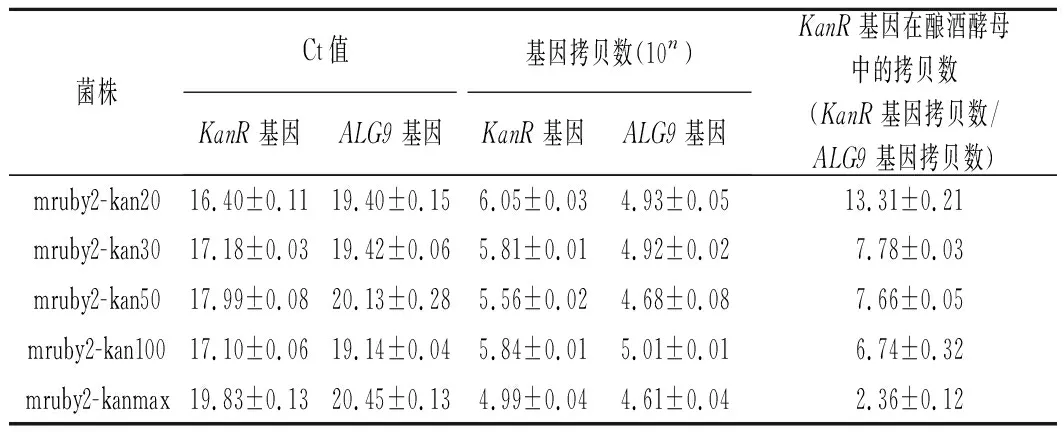
表3 RT-PCR检测KanMX整合盒质粒拷贝数Table 3 RT-PCR to detect the plasmid copy number of truncated KanMX
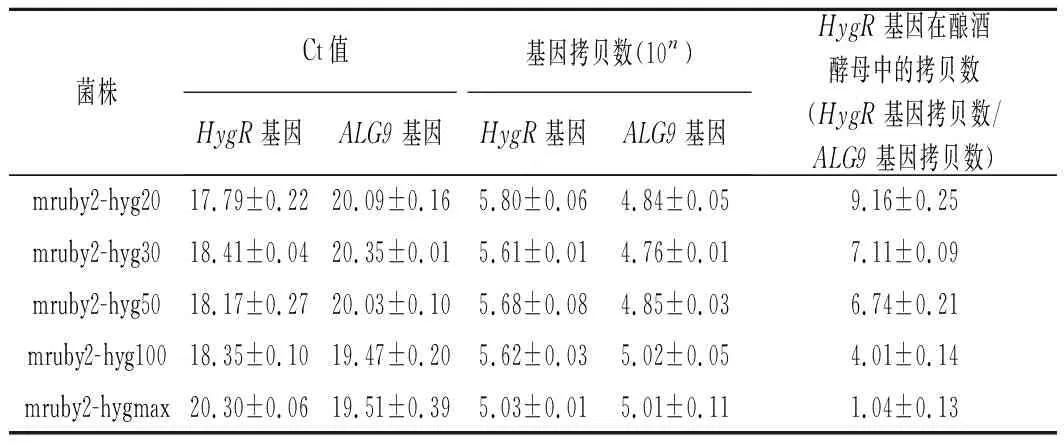
表4 RT-PCR检测HygB整合盒的质粒拷贝数Table 4 RT-PCR to detect the plasmid copy number of truncated HygB
2.5 10,11-dehydrocurvularin的合成与检测
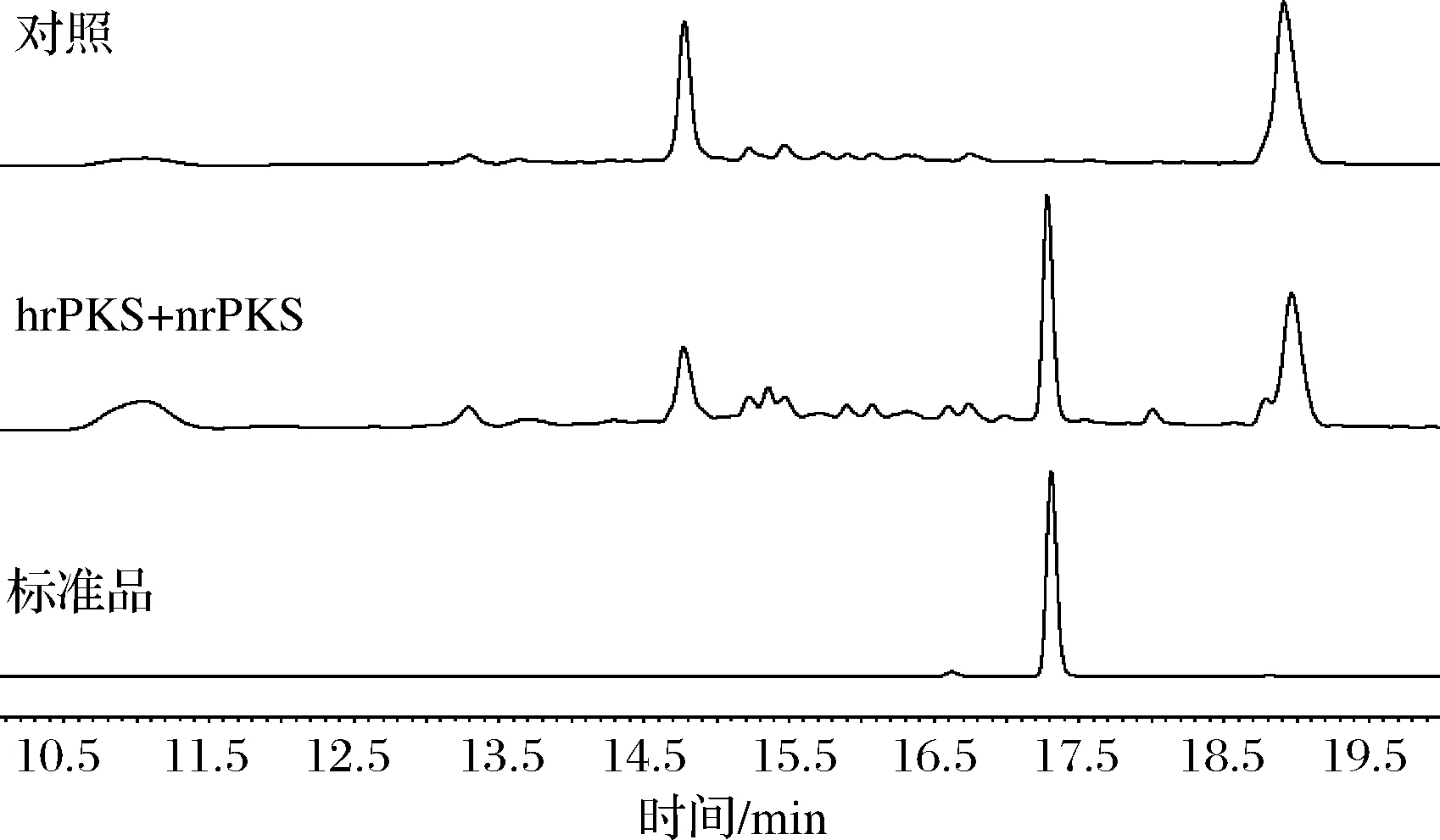
带有质粒pYX003-pXW06F_AtF7_RedPKS、pYX004-pXK30F_AtF7_nrPKS的BJ5464-NpgA菌株与对照菌株在SC培养基发酵2 d的发酵产物经HPLC处理结果如图4所示。在标准品的HPLC结果对比下,表明10,11-dehydrocurvularin成功在酿酒酵母中合成。
2.6 使用优化10,11-dehydrocurvularin合成途径以提高产量
将分别携带1种转录缺陷表达盒与聚酮合酶的两类质粒进行组合转化至BJ5464-NpgA表达,共有5×5即25种组合。每个转化挑取3个单克隆,使用24孔深孔板在SC培养基中进行预培养1 d,以2%转接含300 mg/L G418与300 mg/L潮霉素B的SC培养基中培养2 d,经上述处理方法后,测得其发酵产量如图5所示。
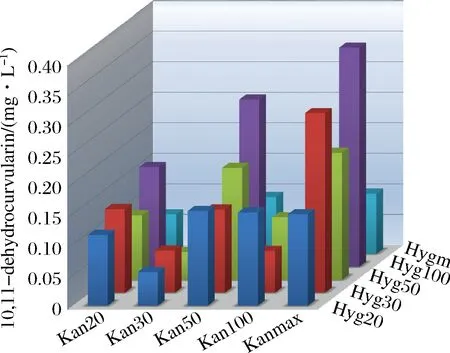
在2种抗生素基因启动子完整的情况下,10,11-dehydrocurvularin产量为0.098 8 mg/L,在使用整合盒的情况下,产量最高的组合能使产量达到0.359 mg/L,是对照组即完整启动子组合的3.63倍。
3 结论与讨论
已有报道质粒拷贝数一定程度与目标蛋白表达量成正相关[20]。本研究显示出所使用的转录结合缺陷抗生素抗性基因整合盒或许能够作为一种提高质粒拷贝数从而增强基因表达元件被开发[13]。其独立性和通用性仍需从2个方面进行考量:拷贝数的变化是否独立于待表达基因、应用到原核生物是否可行。即不同基因携带转录结合缺陷抗生素抗性基因整合盒是否具有相同的增强功能,在不同的宿主内使用这种方法能否达到相同的效果。
异源表达生产有价值的产品有很大的优势,以截短侧耳素为例,截短侧耳素是一种由担子菌产生的抗生素,其在天然宿主中的产量不足以用于遗传改造,但其在米曲霉中的异源表达使效价提高了2 000%,这使得这种重要抗生素首次可持续生产[6]。除了本文使用的调控质粒拷贝数来优化聚酮类化合物产量的方法,也有一些研究是通过优化氧化代谢途径提高产量[1]。氧化,是聚酮类化合物翻译后修饰的重要步骤[3],而大肠杆菌由于缺少一些后修饰功能,不是普适性的聚酮类化合物异源表达宿主[21]。
本课题在酿酒酵母中异源表达来自土曲霉的聚酮合酶合成10,11-dehydrocurvularin,并对该酶的代谢途径进行了优化,提高产量。本研究发现,该表达盒在KanamycinR和HygromycinR基因启动子分别为完整长度和100 bp时异源合成10,11-dehydrocurvularin能获得最大产量,最高产量约为0.359 mg/L。我们可以发现,并非合成酶基因拷贝越多,产量越高,发酵过程中合理的资源利用更有利于目的产物的合成。此研究将有利于今后非天然聚酮化合物的发现及优化生产,随着越来越多的聚酮类化合物的活性被挖掘出来,如何去大量获取这些活性分子是不可缺少的研究方向。
