氮杂环有机催化CO2环加成反应:咪唑环的弱协同效应
2021-07-24王结祥关磊叶松寿郑进保陈秉辉
王结祥,关磊,叶松寿,郑进保,陈秉辉
(1 厦门大学化学化工学院,醇醚酯化工清洁生产国家工程实验室,福建厦门361005;2山东联创产业发展集团股份有限公司,山东淄博255022)
引 言
CO2是一个热力学上相对稳定的分子,需要通过高能化合物才能活化,如环氧化物、二烯、胺类及氢气等。在大力提倡“碳达峰、碳中和”背景下的CO2资源化利用上,作为“原子经济性”的反应,针对CO2环加成反应、CO2加氢还原[1-3]、CO2羧基化[4]等近年来展开了一系列研究。其中,反应能垒相对温和的CO2环加成反应是研究较早、机理相对清晰的一个反应。环氧化物的开环是CO2插入活化的关键,为了提高CO2环加成活性,一般会引入亲核试剂,如引入金属卤化物、烷基季铵盐或构建多氢键协同效应等。
较早研究的是引入金属卤化物(如在离子液体中引入氯化铁[5]、氯化锌[6])、Salen 铝盐[7]及有机锑、铋[8]等,但因为金属卤化物容易脱落,影响催化剂的重复利用,造成金属在溶液中的残留。进一步开展了添加有机类(如烷基铵盐)的催化体系研究,例如双金属水滑石和四丁基溴化铵[9]、全氟三醇化合物和四丁基碘化铵[10]这种多元组分的协同催化实现了CO2环加成的高转化率,并向着温和(室温、1 标准大气压CO2)、高效的方向发展。Zhang 等[11]通过合成咪唑功能化的金属络合物实现了快速的CO2环加成反应(反应时间2 h)。
随着绿色化学的提倡,多元催化体系向着单元催化体系及无金属、无卤素、无溶剂方向发展,形成了一系列多氢键协同催化体系[12],这种氢键包括羧基[13]、羟基[14]、胺基等。Liu等[15]合成了兼具氮杂环卡宾基团和羧基的钳状化合物、Wu等[16]设计了兼具胺基和羟基的Salen 结构化合物、Hao 等[17]合成了兼具三唑和酚羟基的钳状化合物。它们都通过分子内协同效应实现了高效的有机催化CO2环加成,部分也可以在室温、1标准大气压CO2的条件下进行。
在追求温和活化CO2环加成方面,虽然间歇反应中均相催化通常可实现高于非均相催化的活性,但非均相催化在实现连续化反应、快速分离等方面显得更为高效,这也是实现绿色制备的重要一环。Sainz Martinez 等[18]通过将离子液体负载于二氧化硅上,实现了高效的CO2连续转化,但同时,催化剂的连续稳定性因为聚碳酸丙烯酯的生成,在离子液体层团聚而快速失活。一方面,其失活在于单体选择性不高;另一方面,在于无机载体二氧化硅本身的孔结构和比表面性质引发。生成聚合物。Yingcharoen 等[19]通 过pKa量 化 了 羟 基 氢 键 的Brönsted 酸性,并关联CO2环加成反应的催化活性,指出pKa在9~11之间(如苯酚)是比较适宜单体碳酸丙烯酯(PC)的生成。过高的酸性虽然有助于环氧化物开环活化,但同时可能造成产物PC在环加成环节不易离去,进而聚合形成低聚物。Léonard 等[20]通过优化载体合成方法,构建了具有介孔大比表面积的二氧化硅载体,实现了非均相催化剂比相应均相活性中心催化剂更高的活性。相比有机载体如聚苯乙烯(PS)、聚乙烯吡咯烷酮(PVP),无机载体二氧化硅在有机溶剂中溶胀性较差,孔结构不易伸缩,积碳更为明显。而且,这种载体负载型的催化剂普遍存在接枝率不高的问题,为此,应构建基团更为丰富的金属功能化的多孔有机聚合物材料[21-23]。Subramanian 等[24]报道了咪唑功能化的有机聚合物(COP-222)在无助剂、无金属、无溶剂且1 标准大气压CO2下实现了高效的环加成催化。
从多元催化体系到单元多氢键协同的有机催化体系,从高活性的均相催化到绿色的非均相载体构建,CO2活化一直朝着高效、温和、绿色的方向发展。在工业推广方向,对催化剂的要求则更倾向于稳健、简单、可重复性,如无金属、无添加剂、无溶剂等,最好是单组分催化剂,如上述的COP-222。这类反应多数的单组分催化剂为氮杂环,又以咪唑型最普遍,少数报道如方酰胺[25]、吡唑[26]、吡啶等,却很少对比不同氮杂环催化活性上的差异及其机制。为了突显活性组分的催化机制、减少其他组分和孔结构差异等的干扰,本文以聚苯乙烯负载氮杂环进行研究,在相对简化的环境下,引入较为普遍的五元和六元氮杂环,考察其对CO2吸附和活化的差异。
1 实验材料和方法
1.1 材料
交联1% DVB 的氯甲基聚苯乙烯树脂(Merrifield 树脂,PS-CH2Cl,聚苯乙烯中交联1%DVB,2.0 mmol/g)采购自天津南开和成有限公司;CO2气体纯度为99.99%;除特别说明,其他试剂主要采购于国药集团化学试剂有限公司。
1.2 催化剂的合成
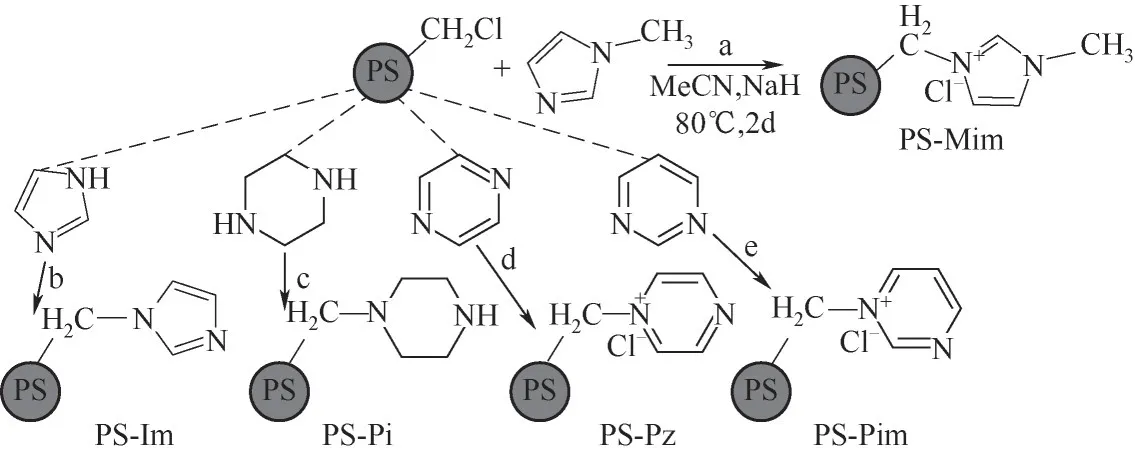
图1 PS-N的合成Fig.1 Synthesis of polystyrene-supported N-heterocycles
1.2.1 合成聚苯乙烯负载的含氮杂环化合物 合成路线见图1。以N-甲基咪唑(Mim)为例:将氯甲基聚苯乙烯(1%DVB,1 g,2.0 mmol Cl含量)溶胀于50 ml乙腈(MeCN)中;恒温50℃搅拌2 h;然后,依次加入Mim(0.4 g,4.9 mmol)和NaH(0.2 g,4.9 mmol),加热到80℃,冷凝回流,恒温搅拌2 d;之后,冷却到室温,依次经过过滤、乙酸乙酯洗涤和甲醇洗涤,产物在110℃下真空干燥4 h。通过元素分析得到产物PS-Mim 上Mim 负载量为1.43 mmol/g。为了消除卤素残留影响,通过充分洗涤后,还需对最后的母液滴加AgNO3溶液,直至无沉淀生成为止。
同样地,将Mim 换成其他含氮杂环,以同样的物质的量进行合成,包括哌嗪(Pi)、吡嗪(Pz)、嘧啶(Pim)和咪唑(Im),并依次命名为PS-Pi(接枝量2.12 mmol/g)、PS-Pz(1.2 mmol/g)、PS-Pim(1.54 mmol/g)、PS-Im(2.09 mmol/g)。
1.2.2 聚苯乙烯咪唑盐螯合氯化锌的合成 氮气气氛下,将PS-Mim(1 g)、ZnCl2(0.15 g, 1.15 mmol)和50 ml CH3CN 混合搅拌,并在80℃冷凝回流72 h之后,过滤、洗涤制得PS-Mim-ZnCl2,如图2(a)所示,并在110℃真空干燥4 h。
1.2.3 PS-Im 上接枝烷基胺的合成 依据文献[27],PS-Im-Ep-TEPA 合成步骤如下:氮气气氛下,将PS-Im(1 g)溶胀于50 ml MeCN 2 h,依次加入NaH(0.08 g, 2 mmol)和 环 氧 氯 丙 烷(Ep,0.16 ml, 2 mmol),室温下反应2 d,制得PS-Im-Ep。然后,添加等物质的量的四乙烯五胺(TEPA,0.4 ml, 2 mmol),60℃下搅拌24 h。产物经过滤、THF洗涤、乙醇洗涤,制得PS-Im-Ep-TEPA,如图2(b)所示,于110℃下真空干燥4 h。
1.3 CO2和环氧丙烷环加成制碳酸丙烯酯
采用300 ml Parr反应釜,投入已知量的PO和催化剂PS-N,装釜。室温下,先用少量的CO2缓慢排空釜内的空气,然后再加入一定压力的CO2气体,稳定10 min 记录下初始的温度、压力。然后,反应釜加热到指定温度,并调节釜搅拌桨转速约为400 r/min 后搅拌反应,通入冷凝水用以冷却搅拌浆。随后每隔2 h 记录一次反应压力和温度。以固定反应时间作为反应终止的标记。反应过程中,不再额外加入CO2(按理想状态气体估算,15 ml PO 对应常温4 MPa CO2,其摩尔比约为1∶2,保证CO2适当过量)。反应结束后,用-15℃的冷肼将反应釜冷却到20℃以下,缓慢放空釜内剩余CO2气体,此时釜内温度会骤降到0℃左右,尽量保证未反应PO 不被带走。过滤掉催化剂(过程中采用密封自然过滤,不使用真空泵抽滤),采用GC和GCMS分析样品组成。
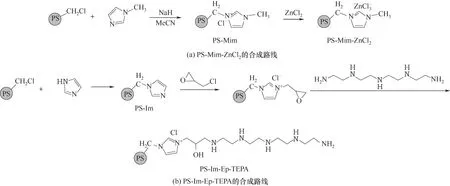
图2 PS-Mim-ZnCl2和PS-Im-Ep-TEPA 的合成路线Fig.2 Synthesis routes of PS-Mim-ZnCl2and PS-Im-Ep-TEPA
1.4 化学分析
1.4.1 产物组成定性、定量分析 产物由气相色谱(Shimadzu GC-2010)分析组成,该色谱装有一根60 m DB-35 毛细管柱,配有FID 检测器。进样口和检测器的温度均保持在260℃,高纯氮(99.999%)为载气,流速1 ml/min,分流比120。程序升温如下:60℃保持2 min,以5℃/min 升温至120℃,再以10℃/min升温至260℃,保持10 min。由于仪器本身稳定性和重现性均较好,采用外标法进行定量分析。产物定性分析由相应的气质联用仪(Shimadzu GCMSQP2010)实现。

1.4.2 其他检测手段 基团接枝量由CHNS 元素分析仪(Vario EL Ⅲ,德国)测定;热稳定性由热重分析仪(SDT-Q600,美国)测定;比表面积和孔结构由物理吸附仪(Micromeritics ASAP2020)测定;CO2脉冲实验由化学吸附仪(Autochem 2920)测定;衰减全反射红外光谱(ATR-IR)和红外光谱(FT-IR)均由红外光谱仪(Bruker VERTEX 70)测定;核磁由液体核磁共振仪(Advance Ⅲ400)测定。
其中,CO2脉冲实验在常压、40℃下,通过数次通入CO2脉冲,让吸附材料充分吸附后,记录每一次脉冲后出口CO2的剩余量,直至出口CO2剩余量基本一致,说明材料吸附达到饱和,以最后一次CO2剩余量作为每次脉冲的投入量,扣除前面几次的剩余量,即得到每次脉冲的吸附量,累加各次CO2的吸附量即得到材料在相应工况下总的CO2吸附量。
另外,用于ATR-IR 和NMR 测定的样品为等物质的量的Mim和PC混合,超声1 h。
PC:1H NMR (400 MHz, CDCl3):δ1.48 (d,J= 6 Hz, 3H), 4.01 (t,J= 8.4 Hz, 1H), 4.54 (t,J= 8.4 Hz,1H),4.80~4.88(m,1H)。
Mim:1H NMR (400 MHz, CDCl3):δ3.58 (d,J= 4 Hz,3H),6.78(s,1H),6.95(s,1H),7.32(s,1H)。
2 实验结果与讨论
2.1 PS-N在CO2环加成上的活性研究
对合成的系列PS-N 进行红外光谱定性分析,如图3(a)所示。以PS-Im 和PS-Mim 为例,从PSCH2Cl 合成PS-Im 的特征信息是668 和1262 cm-1处的C—Cl 特征峰消失,同时出现了咪唑在1668 cm-1的C N 伸缩振动峰。图3(b)通过元素分析计算出氮杂环基团的接枝量。PS-Pi 和PS-Im 拥有较高的负载量,分别为2.12 和2.09 mmol/g,而PS-Pz、PSPim、PS-Mim 接枝量均较低,分别只有1.2、1.54、1.43 mmol/g。可以看出,含仲胺的Pi和Im 比含叔胺的Pz、Pim、Mim 更易接枝,这是因为仲胺的亲核消除反应比叔胺的Menshutkin反应更容易进行。从热稳定性来看[图3(c)],PS-Im的热稳定性明显优于其他4种材料,其热失重曲线只有一段明显的失重区,基团的接枝跟聚苯乙烯骨架热坍塌在同一个温度范围内[28],而PS-Mim的热稳定性较差,在约230℃形成了基团的热解失重,这与仲胺和叔胺的接枝形式有关。热稳定性会影响催化剂在高温、高压、溶剂环境下的重复利用。
进而,将这一系列PS-N 应用于CO2环加成反应,如图4 和图5。五元环结构的PS-Im 和PS-Mim活性(PC 产率分别为97.4%和62.5%)高于六元环的PS-Pi、PS-Pz、PS-Pim。考虑到反应过程中采用同样质量的催化剂,但由于负载量不同,以氮杂环为活性基团,发现PS-Im 和PS-Mim 的转化数(TON,分别为66.6 和62.5)差别不大。可见,氮杂环的分子结构(五元环或六元环)对催化活性有一定影响。另外,作为同分异构体的Pz 和Pim,TON值差别较大。
进一步考察PS-Im 在CO2环加成反应中的条件优化,如表1 所示。温度对活化CO2环加成反应影响很大,随着温度的升高,转化率上升,选择性也略有上升。当温度超过120℃时,选择性略有下降。在单体环状PC和聚碳酸丙烯酯的选择性竞争中,低温有利于开环聚合,高温有利于单体环化[29]。另外,在PO 和CO2摩尔比接近1∶1的情况下,PC 产率仍有95.3%;通过下调反应压力和催化剂用量可以发现,温度120℃、压力3 MPa下,PC产率仍有96.1%。
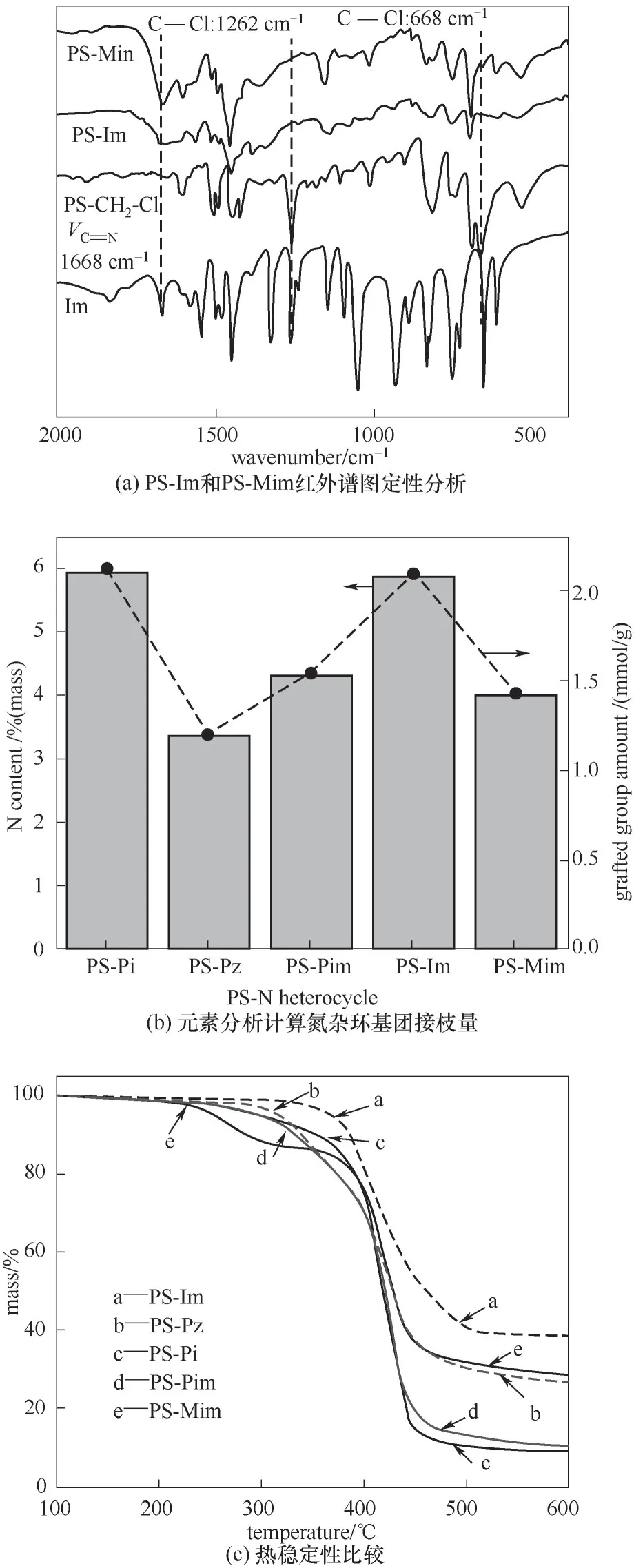
图3 PS-N的定性与定量分析Fig.3 Qualitative and quantitative analysis for PS-N heterocycle series
图4 CO2环加成反应Fig.4 CO2cycloaddition reaction
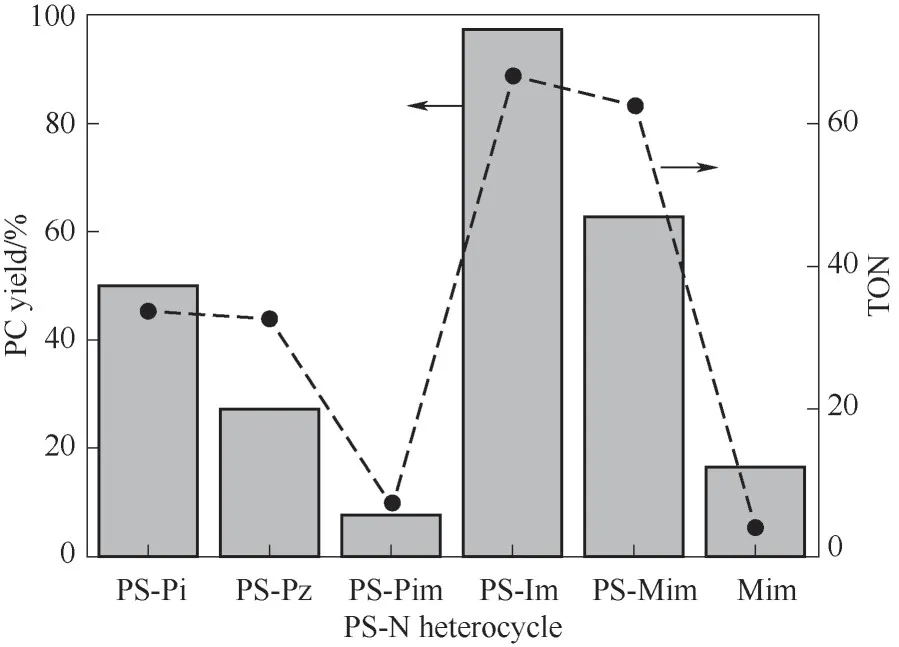
图5 PS-N应用于CO2环加成反应[反应条件:PO 15ml,PCO2 4 MPa(初始压力),催化剂1.5 g(其中,Mim 0.5 g,约为PS-Mim活性组分用量的3倍),T=100℃,t=10 h]Fig.5 CO2 cycloaddition catalyzed by PS-N series
对PS-Im 进行重复利用测试(图6)可知,连续使用4 次未见明显的活性下降,PC 产率和选择性都保持较高水平。不仅在于PS-Im本身的热稳定性和化学稳定性较强,还因为其高选择性也避免了可能的聚合产物在孔内积碳,更在于聚苯乙烯载体在环氧化物里的高溶胀性,从而避免了活性下降的隐忧。这种稳健、粗犷、无金属、无卤素的有机催化剂具有一定的工业应用潜力。
另外,图5中考虑不使用聚苯乙烯非均相载体,由单体Mim 作为均相催化剂,发现其活性反而低于负载型咪唑盐。对PS-Im 进行比表面积测定,其吸附等温线如图7(a)所示。PS-Im比表面积并不大,只有8.153 m2/g,平均孔径在1.89 nm 左右。即使在高温的PO 环境下,PS-Im 的溶胀度大致在3~5 倍左右[30]。可见,PS-Im 或PS-Mim 在比表面积不大的情况下活性甚至比相应的均相活性组分更高,显示出了一定的载体效应。
针对CO2的催化活性对比,考察不同催化剂对CO2的吸附性能,对部分PS-N 系列进行了CO2脉冲测试[图7(b)]。发现PS-Pi 具有更高的CO2吸附量,达1.3×10-2mmol/g。CO2吸附能力依次为:PS-Pi>PS-Mim>PS-Pim>PS-Im,仲胺上的N—H 相比叔胺更有利于CO2的吸附[31]。PS-Im 对CO2的吸附性能最差,但却拥有最高的催化活性。可见,CO2的吸附与活化规律并不一致。在吸附以后,CO2的活化还与三元氧环的开环有很大关系,氮杂环的分子结构(五元环或六元环)对催化活性存在影响。
2.2 PS-N在CO2环加成反应中的机理研究
由于在原位实验中很难捕捉到氮杂环与CO2、PO 的相互作用关系,拟反向思维,从终态氮杂环与PC 的作用关系反向推测氮杂环如何协同PO 开环、活化CO2插入加成的过程。考虑到聚苯乙烯载体作为一个非均相惰性基团不易在均相中引入,拟用惰性的甲基进行替换,即Mim视为PS-Im的替代物,而不是直接用Im 替代[氢可能对反应有影响,如图8(a)]。

表1 PS-Im用于CO2环加成反应的条件优化Table 1 Reaction condition optimization for PS-Im to catalyzed CO2 cycloaddition
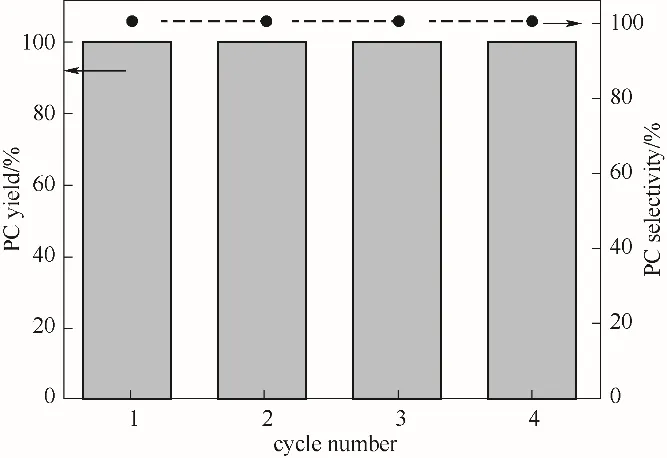
图6 PS-Im在CO2环加成中的重复性测试[反应条件:PO 15 ml,PCO2 3 MPa(初始压力),催化剂0.5 g,T=120℃,t=10 h]Fig.6 Recycling test for PS-Im to catalyzed CO2 cycloaddition
首先,通过核磁分析比较氮杂环与PC间的相互作用,以PC-Mim 和PC-Pz 两种混合液为例,如图9所示。氮杂环的加入使PC 氢谱化学位移向高场移动,且Mim 对PC 作用的化学位移大于Pz 对PC 的作用位移,主要原因在于五元氮杂环的电子云密度比六元环的大,给电子作用使得高场位移更明显,易于质子化和发生亲核反应。
其次,为了考察Mim 和PC 的作用位点,测定其ATR-IR,如图10 所示。从插图中可以看出细微的峰移动。PC-Mim 相对于Mim 和PC 而言,各个有相互作用的峰均发生蓝移现象,其中738、816 cm-1归属于咪唑环上C—H 键的面外弯曲振动,1045 cm-1归属于PC 上C—O 键的伸缩振动,1174 cm-1归属于PC 上C—O—C 键的伸缩振动,1782 cm-1归属于PC上C O 键的伸缩振动,3107 cm-1归属于咪唑环上C—H 或N—H 的对称伸缩振动。结合这些,如图8(b)所示,认为两者的作用主要聚焦于Mim 上的N—CH N跟PC上的O—CO—O间。

图7 PS-N系列的孔结构性质及对CO2的吸附性能Fig.7 CO2adsorption for PS-N heterocycle series
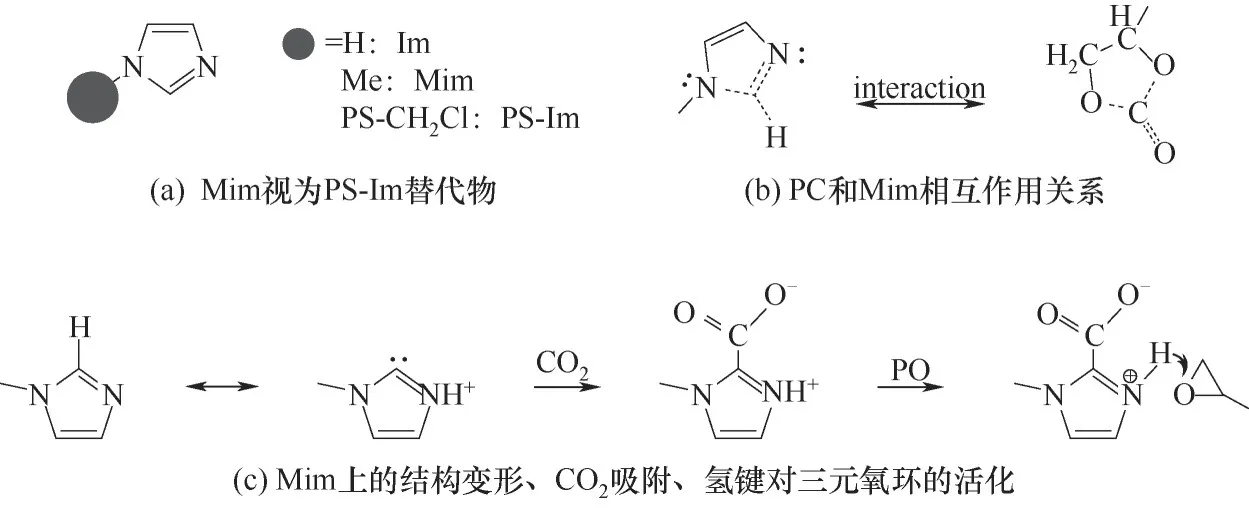
图8 组分间的相互作用示意图Fig.8 The diagram for the interaction of components
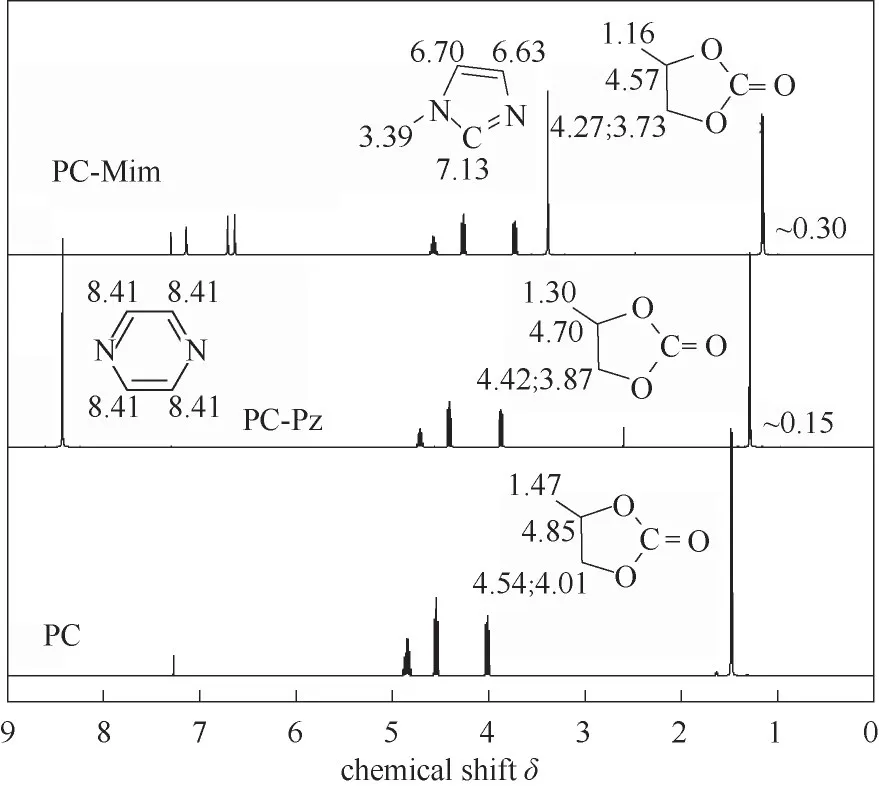
图9 PC-Pz、PC-Mim两种混合液的1H NMR比较Fig.9 1H NMR comparison for PC-Pz and PC-Mim
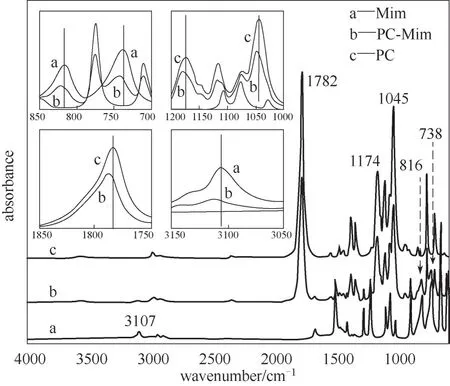
图10 PC-Mim混合液的ATR-IRFig.10 ATR-IR for PC-Mim mixture
如图8(c)所示,电子云密度较大的咪唑C2容易去质子化形成氮杂环卡宾结构,生成带有孤对电子的碳负离子,吸附CO2形成羧酸基咪唑鎓盐[32]。而图7(b)也指明PS-Pi 等对CO2的吸附量高于PS-Im,但活性却不如后者,可见,单独的咪唑环吸附CO2还不足以活化CO2反应,还需要三元氧环的开环协助[33]。五元咪唑环在活性上高于六元环,推测原因在于吸附CO2的同时,具有较高电子密度的五元咪唑环易质子化,结构转换的邻位N—H 键有利于对三元氧环的氧形成氢键作用,协同促进CO2对三元氧环的插入活化,进而推测如图11所示的弱协同作用示意图。
值得注意的是,六元氮杂环的电子云密度较低,如吡啶氮原子上的未共用电子对不参与大π 体系,是吸附CO2的活性位点[34];而五元环的吡唑类离子液体通过两分子吡唑对PO 的氢键协同开环,才促进了CO2的插入活化[26]。可见,不同的氮杂环对CO2环加成反应的活化机制还是有一定差异的。
2.3 改性PS-N在CO2环加成上的应用

图11 PS-Im上咪唑环对反应底物CO2和PO间可能的弱协同催化示意图Fig.11 Plausible schematic diagram of a weak cooperative catalysis between imidazole and substrates(CO2 and propylene oxide)on PS-Im
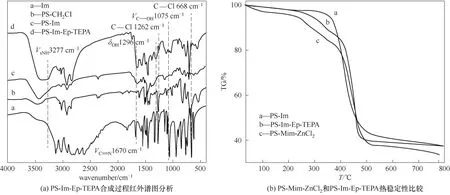
图12 改性PS-N的定性分析Fig.12 Qualitative analysis for the modified polystyrene-supported imidazoles
除了直接考虑氮杂环对CO2的环加成性能,本文也尝试进一步引入金属卤化物和烷基胺进行对比。在PS-Mim 上引入ZnCl2制备了PS-Mim-ZnCl2,在PS-Im 上通过EP 引入TEPA 制备了PS-Im-Ep-TEPA。
对合成的PS-Im-Ep-TEPA 进行红外光谱定性分析。由图12(a)知,从PS-CH2Cl 合成PS-Im(对比谱 线b 和c)的 特 征 信 息 是668 和1262 cm-1处 的C—Cl 特征峰消失,同时出现了咪唑的特征峰即1670 cm-1的C N 伸缩振动峰。从PS-Im 合成PSIm-Ep-TEPA(对比谱线c 和d)后,1075 cm-1归属于—C—OH 的伸缩振动峰,1296 cm-1归属于—OH 的弯曲振动,这是由于PS-Im 接枝上了Ep,Ep 开环出现羟基所致;3277 cm-1归属于—NH 的伸缩振动,表明接枝上了TEPA。
将 合 成 的PS-Mim、PS-Mim-ZnCl2、PS-Im 和PS-Im-Ep-TEPA 应用于CO2环加成反应中,性能如表2 所示。锌盐的引入提高了反应速率,促进了反应转化率的提高,同时,选择性有所下降,可能在于锌盐对PO 的活化作用有利于链增长的进行[35]。同样地,相比于PS-Im,TEPA 中胺基氢键的引入有利于环氧化物开环,加快了反应进程,选择性也有所下降。高的Brönsted 酸性有利于环氧化物的开环,但同时也降低了产物在环化环节的释放,从而造成部分低聚物的产生[19]。可以看出,不管是金属卤化物还是烷基胺的引入,都有助于提高催化活性,加速反应进行,但同时造成选择性下降。另外,图12(b)热重分析显示,TEPA 和ZnCl2的引入降低了聚苯乙烯咪唑盐的耐热性。而且,这种通过离子键负载的金属卤代物在高温、溶剂环境的反应过程中也容易流失。由此,PS-Im 这种制备简单、成本较低、热稳定性高、选择性好的稳健材料更适合于工业推广。
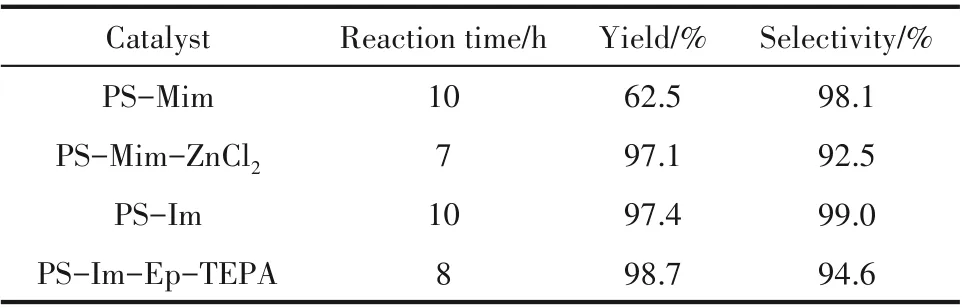
表2 聚苯乙烯负载系列用于CO2环加成反应Table 2 CO2 cycloaddition catalyzed by PS-supported series
3 结 论
胺基被认为是高效的CO2吸附基团,而热力学上稳定的CO2的活化不仅需要高能分子(如环氧化物),还常需要有亲核试剂诱发三元氧环的开环,才能实现CO2的插入活化,这是区分CO2吸附和活化的一个重要分水岭。本文立足于聚苯乙烯负载的有机催化,在不引入金属卤化物或多氢键基团下,考察不同含氮杂环材料在催化CO2环加成中的活性差异,发现五元氮杂环显示出了高于六元氮杂环的催化活性,特别是PS-Im 在重复使用4 次后未发现明显的活性降低,充分体现了PS-Im 较强的热稳定性和化学稳定性。另外,非均相的PS-Mim 和PS-Im甚至比相应的均相活性组分Mim 具有更高的活性,体现了载体的优势效应。
CO2脉冲实验显示PS-Im 的CO2吸附能力反而弱于其他几类氮杂环,推测从CO2的吸附到活化之间还有一道鸿沟,其关键在于PO 的活化。1H NMR指出五元环Mim对PC的作用强于六元环Pz,而ATRIR将这种作用关系聚焦于咪唑环上的N—CH N和即将生成的产物PC 上的O—CO—O,结合文献分析,认为咪唑C2的去质子化实现了CO2的吸附,而质子化的N—H 键对三元氧环的氧形成了氢键作用,两者协同促进了CO2对三元氧环的插入活化。
进一步地,与引入ZnCl2、烷基胺的聚苯乙烯负载型催化剂对比,两者虽然反应速率得以提高,但选择性下降。可见,在无金属、无卤素、无添加剂、无溶剂下,PS-Im 这种简单、廉价、高活性、高选择性、稳定且可回收利用的聚苯乙烯负载氮杂环材料具有一定的工业推广价值。
