表面改性未焙烧TS-1固载金催化丙烯氢氧环氧化反应性能研究
2021-07-24张志华杜威段学志周兴贵
张志华,杜威,段学志,周兴贵
(华东理工大学化学工程联合国家重点实验室,上海200237)
引 言
环氧丙烷(PO)是目前仅次于聚丙烯的第二大丙烯衍生物,主要用于生产聚氨酯、表面活性剂、丙二醇等化工产品[1-2]。目前工业上PO 的生产方法主要有氯醇法和Halcon 共氧化法[2-4]。前者存在环境污染严重、废水和废渣排放量大和设备腐蚀等问题,后者存在工艺流程长、投资大和经济性受联产产品市场影响较大等问题[1-2,5]。过氧化氢液氧化法虽然具有较高的丙烯转化率和PO 选择性,但是该方法以昂贵的双氧水为原料,且双氧水需要现场生产,生产工艺较复杂,再加上后续的产品分离需要用到大量的蒸汽,导致该工艺的生产成本居高不下[5]。Haruta等[6]发现采用沉积-沉淀法(DP法)制备的Au/TiO2催化剂能够在氢气和氧气气氛下催化丙烯直接气相环氧化生成PO,他们认为该反应过程的进行需要金和钛的协同效应,氢气和氧气在纳米金颗粒(<5 nm)上反应生成的HOOH物种溢流到Ti位点上形成Ti—OOH 中间体,然后丙烯再和Ti—OOH 物种发生环氧化反应生成PO[7-9]。与其他工艺相比,丙烯氢氧环氧化制备PO 工艺具有环境友好、操作简单、产物易分离和选择性高等优点,日益受到研究人员的广泛关注,成为当今研究领域的热点[1]。
目前,多种含钛载体(如TS-1、Ti-MCM-41、Ti-SBA-15、Ti-MCM-48、Ti-TUD、Ti-MWW)固载的金催化剂被应用于丙烯氢氧环氧化反应中[10-16]。其中TS-1由于具有丰富的孤立四配位钛物种,将纳米金颗粒固载在TS-1 上制备的Au/TS-1 催化剂显示出优异的催化活性[10-16]。然而,即使是TS-1 固载的纳米金催化剂仍然存在稳定性差的问题,严重制约着丙烯氢氧环氧化工艺的工业化应用[17]。本课题组[18]研究发现反应中产生的焦炭堵塞TS-1 微孔孔道是导致Au/TS-1催化剂快速失活的主要原因。基于该失活机理,采用微孔孔道被模板剂堵塞的未焙烧钛硅分子筛TS-1-B 作为载体,以氢氧化钠为沉淀剂通过DP 法将金颗粒固载在TS-1-B 的外表面。采用该方法制备的Au/TS-1-B 催化剂在30 h 的考评时间内未发现明显的失活现象,显示了较好的稳定性[18]。值得指出的是,TS-1 分子筛在晶化过程中不可避免地会产生一些缺陷而形成Si—OH 等缺陷位硅物种[19],这会导致亲水性的PO 在催化剂表面的羟基上吸附而发生开环、异构及聚合等反应生成导致催化剂失活的焦炭物种[20-22]。因此,若能对TS-1-B进行改性处理来降低表面缺陷位硅羟基的数量将有助于进一步提高催化剂的稳定性。
除含钛载体外,催化剂制备方法也对Au-Ti 双功能催化剂的性能具有重要影响。目前,DP法是制备Au-Ti 双功能催化剂最常用的方法,该方法制备的Au-Ti 双功能催化剂活性较高,但是金的负载效率通常较低(<3%)[17,23],并且制备过程烦琐,大大限制了催化剂的工业放大。此外,该方法制备的催化剂不可避免地会残留碱金属离子[24],这可能会因碱金属离子的残留量难以控制而对Au-Ti协同效应的研究带来干扰。Lu 等[25]发现以尿素为沉淀剂可以将大部分氯金酸固载到TS-1-B 分子筛上,金的负载效率大于90%,远高于传统的DP 法,且该方法制备的Au/TS-1-B 催化剂在丙烯氢氧环氧化反应中表现出优异的性能。因此,采用尿素为沉淀剂的DPU 法不但能够获得较高的金固载效率,还能够排除碱金属离子的干扰,更有利于研究Au-Ti 协同的本质。
采用有机碱溶液对TS-1 进行后处理改性来改善其催化性能已成为催化领域的一个研究热点。研究发现采用四丙基氢氧化铵(TPAOH)溶液对TS-1 分子筛进行二次晶化改性时发生了溶解-再结晶过程,改性后的TS-1上产生了有利于分子扩散的晶内不规则介孔,并且改性处理过程中TS-1结构进一步完善,减少了硅羟基等缺陷位,提高了TS-1 表面疏水性,从而显著提升了TS-1 分子筛的催化性能[26-28]。后续研究还发现采用四丁基氢氧化铵(TBAOH)、哌啶(PI)及四丙基溴化铵(TPABr)与乙胺(EA)的混合溶液对钛硅分子筛进行二次晶化改性处理均有助于改善钛硅分子筛的扩散性能并提高表面疏水性[26-27,29]。目前,采用有机碱溶液对未焙烧钛硅分子筛TS-1-B 进行二次晶化处理后再固载纳米金颗粒催化丙烯氢氧环氧化反应的研究尚未见报道。本文以尿素为沉淀剂采用DPU 法将纳米金颗粒固载于TPAOH 溶液二次晶化改性的TS-1-B上,并以同样方法将纳米金颗粒固载在未改性TS-1-B 上作为参比催化剂,比较研究了这两种催化剂在丙烯氢氧环氧化反应中的催化性能差异,并结合傅里叶变化红外光谱(FT-IR)及高角度环形暗场-扫描投射电子显微镜(HAADF-STEM)等表征以建立催化剂结构和性能之间的关系。此外,本文还研究了二次晶化处理改性TS-1-B 固载金催化剂在丙烯氢氧环氧化反应中的动力学特性。
1 实验材料和方法
1.1 材料
实验用水为超纯水,电阻为18.2 MΩ。吐温-20,梯希爱(上海)化成工业发展有限公司。TPAOH,上海迈瑞尔化学技术有限公司提供,质量分数为25%。正硅酸四乙酯(TEOS),上海麦克林生化科技有限公司提供,纯度> 99%。异丙醇(IPA),上海麦克林生化科技有限公司提供,ACS 光谱级。钛酸四丁酯(TBOT),赛默飞世尔科技(中国)科技有限公司提供,纯度>99%。氯金酸水合物,天津希恩思生化科技有限公司提供,纯度(质量分数)为47.8% Au。尿素,上海泰坦科技股份有限公司,纯度> 99%。氮气(纯度99.995%),氢气(纯度99.99%),氧气(纯度99.99%),丙烯(纯度99.5%),液化空气(昆山)气体科技有限公司。
1.2 未焙烧钛硅分子筛制备与改性处理
按照文献[18]所述方法制备未焙烧钛硅分子筛TS-1-B:首先将2.00 g 吐温-20 滴加到28.60 g 超纯水中,室温磁力搅拌1 h 后逐滴滴加22.60 g TPAOH,滴加完TPAOH 后再磁力搅拌1 h,然后再逐滴滴加40.53 g TEOS,待加入TEOS 搅拌至澄清后再将0.62 g TBOT 与10.00 g 异丙醇混合液缓慢滴加到上述澄清液中,搅拌1 h 后将混合液在80℃下加热除醇5 h,再将母液移入带有聚四氟乙烯内衬的不锈钢水热釜,然后放入170℃的烘箱中晶化48 h。晶化结束后将水热釜从烘箱中取出,待降至室温后将浆液离心、洗涤后置于80℃恒温烘箱中干燥6 h,最终制备出硅钛比为100 的未焙烧钛硅分子筛TS-1-B。
未焙烧钛硅分子筛TS-1-B 的改性处理按如下方法进行[27]: 将TS-1-B 分子筛加入TPAOH 水溶液中,配比为1 SiO2∶0.04 TPAOH∶20 H2O,室温下搅拌成均一浆液后倒入带有聚四氟乙烯内衬的不锈钢水热釜中,然后将水热釜置于恒温烘箱(170℃)中晶化24 h。晶化结束后将水热釜从烘箱中取出,然后在冷水中快速降至室温,再将浆液离心、洗涤后置于80℃恒温烘箱中干燥6 h,即得改性处理的未焙烧钛硅分子筛TS-1-B-TPAOH。
1.3 催化剂制备
按照文献[25,30]所述方法制备Au-Ti 双功能催化剂:先将一定量氯金酸水溶液(0.96 g/L)与1.00 g TS-1-B-TPAOH 加入盛有40 ml 水的烧杯中,再加入0.09 g尿素,避光条件下连续磁力搅拌,并将该悬浊液从室温升温至90℃,继续搅拌6 h 后将浆液置于50 ml 离心管中离心10 min,离心后用40 ml 水洗涤并再次离心,再将所得固体样品置于真空干燥器中,在室温下干燥12 h 后即得Au/TS-1-B-TPAOH催化剂。通过调节氯金酸的加入量可制备不同金负载量的催化剂。Au/TS-1-B催化剂的制备步骤与Au/TS-1-B-TPAOH 催化剂制备步骤相同。其中,0.09% Au/TS-1-B 及0.09% Au/TS-1-B-TPAOH 催化剂在制备时加入1000 µl 氯金酸溶液,0.08% Au/TS-1-B-TPAOH 催化剂在制备时加入850 µl 氯金酸溶液。
1.4 表征
钛硅分子筛的物相分析在Rigaku D/Max2250 VB/PC 衍射仪(Rigaku公司,日本)上完成,测定条件为管电流100 mA,管电压40 kV,入射光源为Cu 靶,Kα射线(λ=0.154056 nm),扫描速率12 (°)/min。在Perkin-Elmer Lambda 35型紫外-可见漫反射光谱仪(Plerkin-Elmer 公司,美国)上表征钛硅分子筛的钛物种,以BaSO4为参比。在Nicolet 6700 型傅里叶变换红外光谱仪(Nicolet 公司,美国)上表征钛硅分子筛的钛物种及硅羟基,钛硅分子筛样品采用KBr 压片。采用扫描电子显微镜(SEM)表征钛硅分子筛的形貌与尺寸大小,样品分析在JSM360LV(JOEL 公司,日本)上进行。在JEM 2100F型高分辨率透射电子显微镜(HR-TEM)(日本JOEL 公司)上分析钛硅分子筛的形貌,加速电压200 kV,仪器点分辨率0.23 nm,线分辨率为0.14 nm。在ASAP 2020 型气体吸附测试仪(Micromeritics 公司,美国)上表征钛硅分子筛的孔容、比表面积等织构性质。在液氮温度(-196℃)下测定样品的氮气吸附-脱附等温线,吸附前样品在170℃下抽真空预处理。采用Brunauer-Emmett-Teller(BET)模型计算样品的比表面积,样品的总孔容由比压为0.99 时所吸附的氮气体积换算得到。采用等离子体-原子发射光谱测定催化剂上金的负载量。测试前将催化剂溶于王水与氢氟酸组成的混合溶液中,制备出含有一定浓度的金溶液后再进行分析。在IRIS 1000 型原子吸收光谱仪(Thermal Elemental 公司,美国)上进行金负载量的测定,分析波长175~1050 nm。采用高角度环形暗场-扫描投射电子显微镜(HADDF-STEM)分析纳米金颗粒的粒径分布及平均粒径。HAADFSTEM 分析在Tecnai G2 F20 S-Twin(FEI 公司,荷兰)上进行,最大加速电压200 kV,放大倍数110 万倍,点分辨率0.24 nm,晶格分辨率0.102 nm。
1.5 催化剂考评
采用常压固定床实验装置(图1)评价催化剂性能。该装置的反应器为电加热的连续流动常压固定床反应器,该反应器由石英管、不锈钢壳层、保温层、加热设备以及中空铜管这几部分组成,其中最外层为不锈钢壳层,保温层位于不锈钢壳层和中空铜管中间,内径约为0.6 mm 的石英管放置于中空铜管中。催化剂装填量为0.15 g(采用筛网孔径0.15 mm 筛分),催化剂装入石英管后通入H2/N2(36%(体积)H2,50 ml/min)混合气体从室温以1℃/min 的升温速率升温至200℃,然后再切换为原料气(C3H6/H2/O2/N2体积比为1∶1∶1∶7,35 ml/min),并将原料气及产物通入气相色谱中进行在线分析。

图1 催化剂考评装置Fig.1 Schematic diagram of the experimental apparatus for propylene epoxidation
使用GC 2060气相色谱对产物进行定性和定量分析。使用FID 检测器和RESTEK 毛细柱(0.35 mm×30 m)分析丙烯、乙醛、PO、丙烯醛、丙醛和丙酮,采用TCD 检测器和TDX-1 填充柱(3 mm×1 m)分析氢气、氧气和二氧化碳。
通过归一化法计算丙烯转化率、产物生成速率和选择性,具体计算公式如下所示。
丙烯转化率:
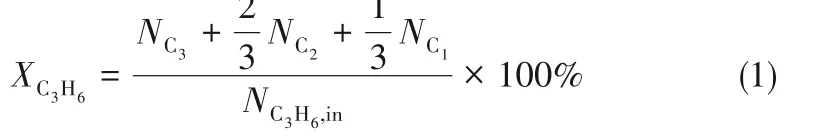
PO生成速率:

PO选择性:
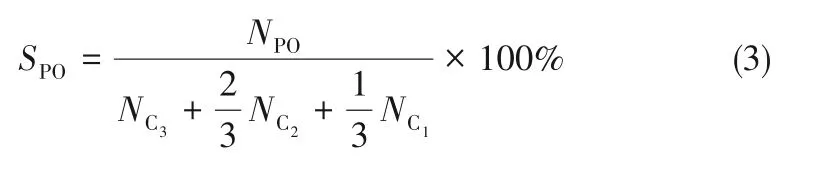
氢效:

2 实验结果与讨论
2.1 TS-1-B-TPAOH与TS-1-B表征
2.1.1 TS-1-B-TPAOH 与TS-1-B 分 子 筛 的 物 相 分析 采用XRD 对TS-1-B 及TS-1-B-TPAOH 分子筛的物相进行了分析,其结果如图2 所示。从图中可 知,TS-1-B 及TS-1-B-TPAOH 分 子 筛 在2θ=7.8°,8.8°,23°,23.9°,24.5°处均出现MFI 结构特征峰[18,31],这说明采用TPAOH 溶液对TS-1-B进行二次晶化改性并未破坏TS-1-B 的拓扑结构[26-27]。此外,与TS-1-B 相 比,TS-1-B-TPAOH 分 子 筛 的MFI 结构特征峰强度明显增强,这说明二次晶化改性提高了TS-1-B的结晶度。这可能是由于TPAOH具有较强的分子筛结构导向能力,在二次晶化处理过程中硅物种发生了溶解-再结晶过程,使得钛硅分子筛结构得到重整、优化,从而提高了TS-1-B 的结晶度[26]。
2.1.2 TS-1-B-TPAOH 与TS-1-B 分 子 筛 中 钛 物 种分析 采用UV-Vis 对TS-1-B 及TS-1-B-TPAOH分子筛中钛物种进行了分析,其结果如图3 所示。从图中可以看出,TS-1-B 及TS-1-B-TPAOH 分子筛均在208 nm 左右出现强烈的电子跃迁信号峰,并且这两个样品在330 nm 处没有观察到明显的锐钛矿型二氧化钛物种吸收峰,这表明所合成的TS-1-B及改性处理后的TS-1-B-TPAOH 分子筛中大部分Ti 以孤立四配位钛物种的形式存在。可见,采用一定浓度的TPAOH溶液对TS-1-B进行二次晶化处理后钛物种的组成并未发生明显变化[18,26,31]。
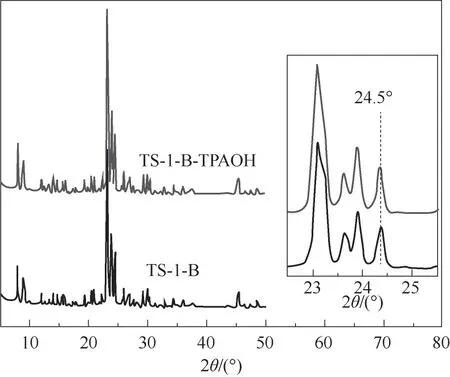
图2 TS-1-B-TPAOH 与TS-1-B的XRD谱图Fig.2 XRD patterns of TS-1-B-TPAOH and TS-1-B

图3 TS-1-B-TPAOH 与TS-1-B的UV-Vis图Fig.3 UV-Vis spectra of TS-1-B-TPAOH and TS-1-B
2.1.3 TS-1-B-TPAOH 与TS-1-B 分 子筛中硅物种分析 TS-1-B 及TS-1-B-TPAOH 分子筛的FT-IR谱图如图4 所示。从图中可以明显看出,TS-1-B 及TS-1-B-TPAOH 分子筛均在960 cm-1处出现Si—O—Ti 的不对称伸缩振动特征峰[18,32],该结果进一步证明钛原子进入了分子筛骨架,并且TS-1-B 及二次晶化后的TS-1-B-TPAOH 分子筛在960 cm-1处的特征吸收峰强度并没有明显的差异,说明二次晶化改性并没有改变TS-1-B 分子筛表面孤立四配位钛的含量。此外,TS-1-B 及TS-1-B-TPAOH 分子筛均在550 cm-1处出现MFI 型分子筛五元环的特征峰[18,33],该结果进一步表明二次晶化改性处理后制备的TS-1-B-TPAOH 分子筛仍具有典型的MFI 结构[26-28],这与XRD 结果相一致。另外,TS-1-B 在约1630 cm-1和3450 cm-1处出现两个特征吸收峰,其中前者归属为氢键硅羟基特征峰,后者归属为氢键硅羟基特征峰及吸附于硅羟基上水分子的特征峰[34-36]。然而,二次晶化改性处理后制备的TS-1-B-TPAOH 分子筛在1630 cm-1和3450 cm-1处几乎观察不到明显的特征峰存在,这可能是由于二次晶化改性处理后TS-1-B 的结构得到了重整、优化,从而降低了缺陷位硅羟基的数量[26-28]。

图4 TS-1-B-TPAOH 与TS-1-B的FT-IR谱图Fig.4 FT-IR spectra of TS-1-B-TPAOH and TS-1-B
2.1.4 TS-1-B-TPAOH与TS-1-B分子筛的形貌
TS-1-B 与TS-1-B-TPAOH 分子筛的SEM 图如图5所示。从图中可以看出,改性处理前后钛硅分子筛颗粒大小并未发生明显的变化,且两个样品颗粒比较均匀并且均呈类球形,并不存在无定形物质。此外,从HR-TEM 图(图6)可以看出,相比于未改性的TS-1-B 样品[图6(a)],改性处理后TS-1-B-TPAOH分子筛[图6(b)]部分颗粒出现了一些不规则的缝隙。这些缝隙可能是二次晶化过程中部分由纳米颗粒堆积形成的TS-1-B进行重新组装后形成的。
2.1.5 TS-1-B-TPAOH 与TS-1-B 分 子 筛 的 织 构 性质 采用N2物理吸附对TS-1-B 与TS-1-B-TPAOH分子筛的织构性质进行了研究。根据脱附分支数据计算的TS-1-B 与TS-1-B-TPAOH 分子筛BJH 孔径分布如图7 所示。从图中可以看出,相比于未改性TS-1-B 分子筛,采用TPAOH 改性的TS-1-BTPAOH 分子筛在3~20 nm 之间显示出较宽的介孔分布,这说明采用TPAOH 二次晶化改性TS-1-B 分子筛后产生了一些介孔。

图5 TS-1-B与TS-1-B-TPAOH 的SEM图Fig.5 SEM images of TS-1-B and TS-1-B-TPAOH

图6 TS-1-B与TS-1-B-TPAOH 的HR-TEM图Fig.6 HR-TEM images of TS-1-B and TS-1-B-TPAOH

图7 TS-1-B与TS-1-B-TPAOH 的孔径分布Fig.7 Pore size distributions of TS-1-B and TS-1-B-TPAOH

表1 TS-1-B与TS-1-B-TPAOH分子筛的织构性质Table 1 The structural properties of TS-1-B and TS-1-B-TPAOH
TS-1-B 与TS-1-B-TPAOH 分子筛的比表面积、微孔孔容及总孔容如表1所示。从表中可知,二次晶化改性处理后制备的TS-1-B-TPAOH 分子筛与未改性处理的TS-1-B 分子筛的微孔孔容相差不大,且远小于传统焙烧后的钛硅分子筛TS-1[18,37],说明二次晶化改性处理后的TS-1-B-TPAOH 分子筛微孔孔道仍然被模板剂TPA+所堵塞。这可能是由于填充于未焙烧TS-1 微孔孔道中的TPA+分子良好的稳定性对TS-1-B 分子筛的微孔孔道具有保护作用,从而避免了二次晶化改性处理过程中强碱性TPAOH 溶液对TS-1-B 微孔孔道的腐蚀。此外,从表中还可以看出,二次晶化改性处理后的TS-1-BTPAOH 分子筛的总孔容要高于TS-1-B 分子筛,这也与TS-1-B-TPAOH 分子筛存在一些介孔这一结果相一致。
2.2 TS-1-B-TPAOH 与TS-1-B 分子筛固载金催化剂性能考评
2.2.1 不同载体固载金催化剂上金负载量分析
表2 为TS-1-B-TPAOH 及TS-1-B 固载金催化剂上金负载量的测定结果。从表2 可知,以TS-1-BTPAOH 及TS-1-B 为载体、采用尿素为沉淀剂通过DPU 法制备的金催化剂上金的负载效率均在90%以上,这与文献报道结果[25]相同。

表2 Au/TS-1-B与Au/TS-1-B-TPAOH催化剂上金负载量Table 2 Au loadings of Au/TS-1-B and Au/TS-1-B-TPAOH catalysts
2.2.2 不同载体固载金催化剂上金颗粒粒径分析
图8 为不同金负载量的Au/TS-1-B-TPAOH 催化剂及0.09% Au/TS-1-B 催化剂的HAADF-STEM 图。从图中可以看出,0.09%Au/TS-1-B-TPAOH 催化剂与0.09%Au/TS-1-B 催化剂上纳米金颗粒的粒径分布相似且平均粒径均为2.6 nm 左右。此外,固载量较低的0.08% Au/TS-1-B-TPAOH 催化剂上纳米金颗粒的平均粒径(约2.5 nm)也与0.09% Au/TS-1-B催化剂的相近,这说明在金负载量相差不大时固载于TS-1-B-TPAOH 与TS-1-B 分子筛上的纳米金颗粒粒径没有明显差异。
2.2.3 催化剂性能考评 不同金负载量Au/TS-1-B-TPAOH 催化剂与0.09% Au/TS-1-B 催化剂在丙烯氢氧环氧化反应中的丙烯转化率和PO 生成速率随时间的变化关系如图9 所示。从图中可知,0.09%Au/TS-1-B 催化剂在诱导期之后的丙烯转化率和PO生成速率随着反应的进行逐渐降低,约30 h后其丙烯转化率和PO 生成速率随着反应的进行不再出现明显的降低,表明该催化剂在30 h 后达到稳态。0.09% Au/TS-1-B-TPAOH 催化剂在诱导期之后的丙烯转化率与PO 生成速率随反应时间变化的曲线可分为两个阶段,其中,该催化剂的丙烯转化率与PO 生成速率在6~20 h 内基本保持不变,之后该催化剂上丙烯转化率与PO 生成速率略有降低但很快就达到稳态,并且该催化剂在稳态时的活性明显高于0.09%Au/TS-1-B 催化剂。此外,0.08%TS-1-B-TPAOH 催化剂在诱导期结束后的丙烯转化率和PO 生成速率随着反应的进行也没有出现明显的降低现象,并且达到稳态时的丙烯转化率和PO 生成速率虽低于同样处于稳态时的0.09% Au/TS-1-B-TPAOH 催化剂,但其活性高于稳态时的0.09%Au/TS-1-B 催化剂。上述催化剂考评结果显示,相比于0.09% Au/TS-1-B 催化剂,0.09% Au/TS-1-BTPAOH 与0.08%Au/TS-1-B-TPAOH 催化剂显示出明显提高的稳定性及稳定的丙烯转化率和PO 生成速率。
先前研究发现,在Ti—OH 活性中心上生成的PO 在相邻Ti—OH 或相邻Si—OH 的作用下能够发生开环反应形成难以脱附的双配位丙氧基物种,该物种可进一步转化为强吸附的羧酸盐(如甲酸盐)和碳酸盐等碳沉积物[20,38-39],这是导致Au-Ti 双功能催化剂失活的主要原因[20,33-39]。研究还发现,Au-Ti双功能催化剂表面形成的双配位丙氧基物种及强吸附的羧酸盐(如甲酸盐)和碳酸盐物种通常在2800~3100 cm-1的红外波长范围内出现C—H 键的伸缩特征峰[20,38-39]。因此,为比较未改性TS-1-B 与二次晶化改性TS-1-B-TPAOH 固载金催化剂上碳沉积物含量及PO 脱附行为的差异,分析了反应后0.09% Au/TS-1-B 与0.08% Au/TS-1-B-TPAOH 催化剂的FT-IR 谱图,其结果如图10 所示。从图中可以看出,反应后0.08% Au/TS-1-B-TPAOH 催化剂与0.09% Au/TS-1-B 催化剂均在2800~3100 cm-1范围内出现了三个归属为双配位丙氧基物种与强吸附羧酸盐(如甲酸盐)和碳酸盐物种C—H 键的伸缩振动特征峰,并且反应后0.08%Au/TS-1-B-TPAOH催化剂上双配位丙氧基物种与强吸附羧酸盐(如甲酸盐)和碳酸盐物种C—H 键的伸缩振动特征峰强度明显低于反应后0.09% Au/TS-1-B 催化剂,这预示着前者在反应过程中表面生成的双配位丙氧基物种与强吸附羧酸盐(如甲酸盐)和碳酸盐物种较少。此外,反应后0.08% Au/TS-1-B-TPAOH 催化剂在1630 cm-1及3450 cm-1处归属于氢键硅羟基及吸附在分子筛硅羟基上水分子的特征峰强度要低于0.09% Au/TS-1-B 催化剂,这也表明二次晶化改性TS-1-B-TPAOH 固载金催化剂的表面疏水性要高于未改性TS-1-B 固载金催化剂。因此,结合双配位丙氧基物种与强吸附羧酸盐(如甲酸盐)和碳酸盐物种的形成机理及二次晶化改性TS-1-BTPAOH 固载金催化剂与未改性TS-1-B 固载金催化剂上C—H 键伸缩振动特征峰和硅羟基特征峰强度的差异可以推测,相比于未改性TS-1-B 固载的金催化剂,二次晶化改性TS-1-B-TPAOH 固载的金催化剂表面硅羟基含量较低,这有利于促进PO 的脱附,导致催化剂表面生成的双配位丙氧基物种及碳沉积物较少,这可能是0.08% Au/TS-1-B-TPAOH催化剂相比于0.09%Au/TS-1-B 催化剂表现出较高的稳定性及稳定的丙烯转化率和PO 生成速率的原因之一。此外,考虑到TS-1-B-TPAOH 的比表面积(24 m2/g)要高于TS-1-B(15 m2/g)。因此,0.08%Au/TS-1-B-TPAOH 催化剂上单位面积的碳沉积物含量也要低于0.09% Au/TS-1-B 催化剂,这可能是0.08%Au/TS-1-B-TPAOH 催化剂相比于0.09%Au/TS-1-B催化剂显示出较高稳定性的另一个原因。

图8 0.09%Au/TS-1-B、0.09%Au/TS-1-B-TPAOH 与0.08%Au/TS-1-B-TPAOH 催化剂HAADF-STEM 图与相应的粒径分布Fig.8 HAADF-STEM images and Au particle size distribution of 0.09%Au/TS-1-B,0.09%Au/TS-1-B-TPAOH and 0.08%Au/TS-1-B-TPAOH catalysts

图9 不同载体固载金催化剂上丙烯转化率与PO生成速率随时间的变化关系(其中,0.09%Au/TS-1-B催化剂的PO生成速率数据来自本课题组先前报道工作[30])Fig.9 Propylene conversion and PO formation rate over 0.09%Au/TS-1-B,0.09%Au/TS-1-B-TPAOH and 0.08%Au/TS-1-BTPAOH catalysts as a function of time-on-stream(the PO formation rate of 0.09%Au/TS-1-B was taken from previous work[30])

图10 反应后0.09%Au/TS-1-B与0.08%Au/TS-1-BTPAOH催化剂的FT-IR谱图Fig.10 FT-IR spetra of spent 0.09%Au/TS-1-B and 0.08%Au/TS-1-B-TPAOH catalysts
催化剂考评结果显示(图9),0.08% Au/TS-1-B-TPAOH 催化剂在诱导期之后的丙烯转化率及PO生成速率低于0.09% Au/TS-1-B-TPAOH 催化剂,但其稳定性要优于金固载量较高的0.09% Au/TS-1-B-TPAOH 催化剂。这可能是由于该催化剂上PO生成速率低于0.09% Au/TS-1-B-TPAOH 催化剂,此时0.08% Au/TS-1-B-TPAOH 催化剂上PO 生成与脱附的良好匹配使得该催化剂相比于0.09% Au/TS-1-B-TPAOH 催化剂显示出进一步提高的稳定性。因此,根据上述分析也可推测,协调Au-Ti双功能催化剂上PO 的生成与脱附是提高Au-Ti 双功能催化剂稳定性的关键所在。
除稳定性外,PO 选择性也是评价Au-Ti 双功能催化剂性能高低的重要指标之一。先前研究发现Ti活性位点上生成的PO能够吸附于相邻Si—OH上并后续发生开环及异构等反应生成副产物[21,33-36],由此可见,促进Ti 活性位点上PO 的脱附有助于提高PO 选择性。Au/TS-1-B-TPAOH 催化剂与Au/TS-1-B 催化剂在稳态时PO 选择性如图11 所示。从图中可知,0.09%Au/TS-1-B-TPAOH 催化剂在稳态时的PO 选择性略微高于0.09%Au/TS-1-B 催化剂,这可能是由于0.09% Au/TS-1-B-TPAOH 催化剂表面硅羟基数量较少,这有利于促进PO 在催化剂表面的脱附,因此该催化剂在稳态时丙烯转化率明显高于稳态时0.09%Au/TS-1-B 催化剂的情况下,其PO选择性仍略高于后者。此外,0.08% Au/TS-1-BTPAOH 催化剂在稳态时的PO 选择性明显高于稳态时的0.09% Au/TS-1-B-TPAOH 催化剂。考虑到0.08%Au/TS-1-B-TPAOH 催化剂上PO 的生成速率低于0.09%Au/TS-1-B-TPAOH 催化剂,此时0.08%Au/TS-1-B-TPAOH 催化剂上PO 的生成与脱附可能实现了较好的匹配,这可能是该催化剂相比于0.09%Au/TS-1-B-TPAOH 催化剂具有较高PO 选择性的原因之一。此外,0.08%Au/TS-1-B-TPAOH 催化剂在稳态时的PO 选择性要明显高于稳态时的0.09%Au/TS-1-B 催化剂。考虑到该0.08%Au/TS-1-B-TPAOH 催化剂的表面疏水性要高于0.09%Au/TS-1-B 催化剂,这有助于促进PO 在催化剂表面的脱附,这可能是该催化剂在具有较高PO 生成速率与丙烯转化率情况下还显示出较高PO 选择性的主要原因。

图11 不同载体固载金催化剂在稳态时的PO选择性与氢效(0.09%Au/TS-1-B催化剂的PO选择性和氢效数据来自本课题组先前报道工作[30])Fig.11 PO selectivity and hydrogen efficiency over 0.09%Au/TS-1-B,0.09%Au/TS-1-B-TPAOH and 0.08%Au/TS-1-BTPAOH catalysts at steady state(the PO selectivity and hydrogen efficiency of 0.09%Au/TS-1-B was taken from previous work[30])
氢效也是评价Au-Ti双功能催化剂性能的重要指标之一。Au/TS-1-B-TPAOH 催化剂与Au/TS-1-B 催化剂在稳态时的氢效如图11 所示。从图中可知,0.09% Au/TS-1-B-TPAOH 与0.08% Au/TS-1-B-TPAOH 催化剂在稳态时的氢效均明显高于0.09% Au/TS-1-B 催化剂,这可能是由于以TS-1-B-TPAOH 分子筛为载体固载的金催化剂表面疏水性较高,这有助于促进PO 在催化剂表面的脱附,从而促进了纳米金颗粒上氢气与氧气反应生成的HOOH 物种与Ti 活性位点反应生成更多的Ti—OOH活性中间体,进而提高了氢气利用效率[21,33-36]。
2.2.4 反应温度影响 反应温度也对Au-Ti双功能催化剂催化丙烯氢氧环氧化反应具有重要影响。0.08% Au/TS-1-B-TPAOH 催化剂上反应温度对丙烯氢氧环氧化反应性能的影响如图12 所示。从图中可以看出,丙烯的转化率和PO 生成速率随着反应温度的升高而提高,但是PO 选择性和氢效却随着反应温度的升高而降低。产生这种现象的主要原因可能是随着反应温度的升高,氢气和氧气在纳米金颗粒上生成HOOH 物种的速率和丙烯环氧化的速率均增加,因此使得丙烯转化率和PO 生成速率也随反应温度的升高而提高。然而HOOH 物种的稳定性不高,较高的反应温度会提高该物种的分解速率,导致大量HOOH 物种在高温下发生无效分解,并且HOOH 物种分解量的增加也提高了非选择性氧化反应生成副产物的速率,从而导致PO 选择性和氢效随反应温度的升高而降低[32,40-41]。
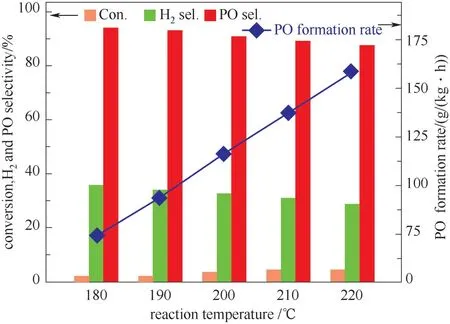
图12 反应温度对0.08%Au/TS-1-B-TPAOH 催化剂性能的影响Fig.12 Catalytic performance of 0.08%Au/TS-1-B-TPAOH catalyst as a function of reaction temperature
0.08% Au/TS-1-B-TPAOH 催化剂上副产物选择性与反应温度的关系如图13 所示。从图中可以看出,该催化剂上生成的副产物有乙醛、丙烯醛、丙醛、丙酮和二氧化碳,其中丙醛是主要副产物。此外,从图中可以看出,随着反应温度的升高,各个副产物的选择性急剧增加,这预示着升高反应温度更有利于副产物的生成[32,40,42]。因此,对于丙烯氢氧环氧化反应而言,提高反应温度反而不利于获得较高的PO选择性。

图13 0.08%Au/TS-1-B-TPAOH 催化剂上反应温度对副产物选择性的影响Fig.13 By-products selectivity of 0.08%Au/TS-1-B-TPAOH catalyst with different reaction temperature
主/副产物生成速率与反应温度的关系如图14所示,通过Arrhenius 方程拟合得到的主/副产物表观活化能如表3 所示。从表中可以看出,生成各副产物的活化能均高于生成主产物PO 的活化能,这表明副产物的生成对反应温度更加敏感[32,40]。此外,0.08% Au/TS-1-B-TPAOH 催化剂上主/副产物的生成活化能位于文献报道的Au-Ti 催化剂上生成主/副产物活化能的范围内,说明本文制备的催化剂上丙烯氢氧环氧化反应与文献报道的催化剂遵从相同的反应机理[32,40,43]。

图14 0.08%Au/TS-1-B-TPAOH 催化剂上主/副产物生成速率与温度关系Fig.14 The relationship between the formation rate of PO/byproducts and temperature over 0.08%Au/TS-1-B-TPAOH catalyst
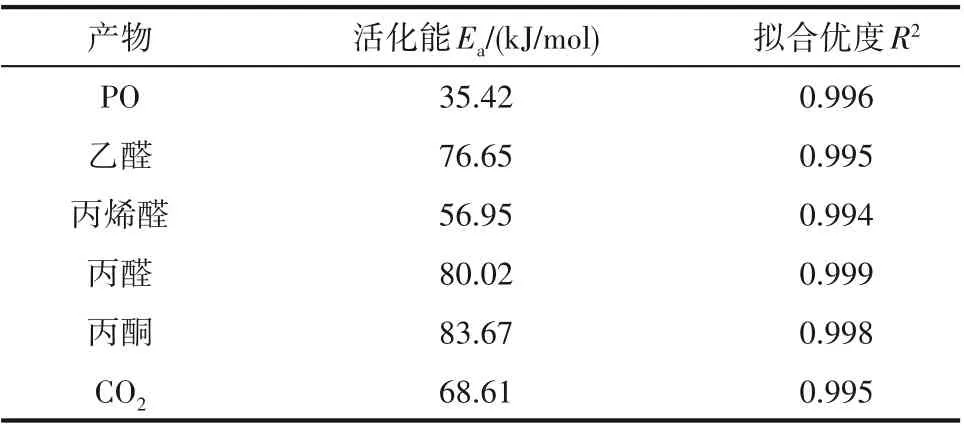
表3 主/副产物表观活化能Table 3 Apparent activation energy of each by-product by linear regression
3 结 论
本文采用TPAOH对TS-1-B进行二次晶化处理制备了TS-1-B-TPAOH 分子筛,对比研究了DPU法制备的Au/TS-1-B催化剂与Au/TS-1-B参比催化剂在丙烯氢氧环氧化反应中催化行为的差异,并结合FT-IR、HAADF-STEM 等表征手段阐释了Au/TS-1-B-TPAOH 与Au/TS-1-B 催化剂结构与性能之间的关系。在此基础上进行了Au/TS-1-B-TPAOH 催化剂的动力学特性分析。结论如下。
(1)TS-1-B 和TS-1-B-TPAOH 分子筛表征结果表明二次晶化改性提高了母体TS-1-B 分子筛的结晶度,减少了分子筛表面缺陷位硅羟基的数量,从而显著提高了TS-1-B分子筛的表面疏水性。
(2)Au/TS-1-B-TPAOH 催化剂相比于Au/TS-1-B催化剂表现出显著提高的稳定性及稳定的转化率、PO 生成速率、PO 选择性和氢效,并且反应后Au/TS-1-B-TPAOH 催化剂上生成的双配位丙氧基物种与甲酸盐等碳沉积物较少,这可归因于二次晶化改性TS-1-B 分子筛显著降低了表面硅羟基数量,这有利于抑制PO 吸附于硅羟基上并发生开环、异构等反应生成双配位丙氧基物种与碳沉积物,从而提高了Au/TS-1-B-TPAOH 催化剂的稳定性与活性。
(3)动力学分析结果显示,提高反应温度虽有利于提高丙烯转化率和PO 生成速率,但会显著降低PO选择性和氢效。生成PO的活化能明显低于生成各副产物的活化能,这表明副反应对反应温度更加敏感。
符 号 说 明
M——摩尔质量,g/mol
m——催化剂质量,g
N——摩尔流量,mol/h
r——生成速率,g/(kg·h)
S——选择性,%
X——转化率,%
下角标
in——进口
out——出口
