利用WGCNA鉴定花生主茎生长基因共表达模块
2021-07-19刘兆新赵继浩赖华江潘小怡李向东杨东清
汪 颖 高 芳 刘兆新 赵继浩 赖华江 潘小怡 毕 晨 李向东 杨东清
利用WGCNA鉴定花生主茎生长基因共表达模块
汪 颖 高 芳 刘兆新 赵继浩 赖华江 潘小怡 毕 晨 李向东 杨东清*
山东农业大学农学院/ 作物生物学国家重点实验室, 山东泰安 271018
以不同主茎高花生品种为材料, 利用转录组测序技术分析茎秆转录组基因表达的异同, 并结合加权基因共表达网络分析(WGCNA), 深入挖掘与主茎生长相关基因, 深入认识花生茎秆形态建成的分子机制。结果表明, 矮秆型Df216与高秆型花育33号相比较共有5872个差异基因; Df216与中间型山花108相比较共有6662个差异基因, 这些差异基因涉及细胞壁和次生细胞壁的生物起源及调控过程、苯丙烷生物合成及代谢过程、木质素生物合成过程、纤维素合酶活性等分子功能。WGCNA鉴定到5个与主茎高呈极显著相关的共表达模块。编码咖啡酰辅酶A-O-甲基转移酶、转录因子ATAF2、WAT1、GDSL脂肪酶等基因是模块内的核心基因。通过筛选权重值构建核心基因的局部网络发现, Grey60模块的核心基因与编码莽草酸香豆酯/奎酸酯3’-羟化酶、4-香豆酸辅酶A连接酶、羟基肉桂酰辅酶A莽草酸/奎尼酸羟基肉桂酰转移酶、以及快速碱化因子、锌指蛋白、类COBRA蛋白等基因有较高互作网络关系; Brown模块核心基因则与编码b-1,4-半乳糖基转移酶、果胶乙酰酯酶、类受体丝氨酸/苏氨酸蛋白激酶、富含亮氨酸重复序列的伸展蛋白等基因有较高互作网络关系。相关模块与核心基因的挖掘以及基因生物学功能和互作网络的解析有助于揭示花生主茎生长的遗传基础。
花生; 主茎高; 转录组; 加权基因共表达网络; 核心基因
株型是指作物的形态特征及其空间的排列方式,不同株型显著影响作物的产量形成[1]。花生株型结构由主茎高、侧枝长以及分枝夹角等组成, 其中主茎高是决定株型指数的重要因子之一。当前我国主推花生品种多为直立紧凑型, 其双仁率高, 结果集中, 适宜密植栽培, 但因其主茎较高, 易倒伏, 农业生产中需喷施外源生长调节剂调控株高[2]。因此开展花生株高调控的分子机理研究, 对花生品种选育以及栽培调控具有重要理论参考意义。
前人对小麦、水稻等禾谷类作物株高相关基因如、, 以及、、等转录因子基因开展了大量研究, 这些基因多与赤霉素、油菜素内酯等植物激素合成代谢和其信号转导相关[3-8]。近年来组学技术和生物信息学迅速发展, 在基因发掘与功能鉴定、作物品种改良、作物抗逆性等方面提供了新思路和新方法[9-12]。Wang等[13]通过转录组与精细定位联合分析确定了控制油菜株高的新候选基因。马娟等[14]通过转录组与加权基因共表达网络分析(weighted correlation network analysis, WGCNA)结合挖掘了与玉米株高显著相关的核心基因及其互作网络。巨飞燕等[15]利用WGCNA筛选与棉花果枝伸长相关的模块发现, JAZ类基因是控制节间伸长的关键基因。前人利用数量性状定位和全基因组关联分析方法对花生茎高遗传开展了相关研究。Huang等[16]利用花生重组自交系(recombinant inbred lines, RIL)群体定位了8个与主茎高相关的QTL位点。Zhang等[17]检测到13个与主茎高显著相关SNPs。针对花生株型、茎高等方面的研究已有部分报道[18-20], 但关于挖掘主茎生长调控的关键基因及其互作网络的研究还尚缺乏。本研究以高秆型花育33号、中间型山花108和矮秆型Df216为材料, 在茎秆生长期考察不同品种茎秆生长动态差异, 利用转录组测序技术分析不同茎高植株茎秆转录组异同, 并结合WGCNA划分基因模块, 深入挖掘与主茎生长相关的基因及调控网络, 以期为花生主茎的生长调控机制研究和花生株型育种提供依据。
1 材料与方法
1.1 试验设计
试验于2019年花生生长季在山东省泰安市山东农业大学农学试验农场(36°9′N, 117°9′E)进行。试验地耕层(0~20 cm)土壤含有机质13.06 g kg-1、全氮10.70 g kg-1、碱解氮70.31 mg kg-1、速效磷42.09 mg kg-1、速效钾57.50 mg kg-1。供试品种材料为直立高秆品种花育33号、直立中间型品种山花108、矮秆花生Df216。小区面积为10.8 m2(4.0 m × 2.7 m), 随机排列。播种前施复合肥750 kg hm-2。于2019年5月8日起垄覆膜穴播方式种植(每个小区起3垄, 每垄种植2行, 垄面行距0.3 m), 每穴2株, 穴距16 cm。2019年9月11日收获。为与自然条件下的高秆(花育33号)、矮秆品种(Df216)做对比, 进一步明确茎秆生长转录组基因表达特点, 选取中间型品种山花108设置2个外源激素处理: 出苗后55 d, 以不喷施为对照, 对山花108喷施浓度为150 mg L-1的多效唑(PP333)和100 mg L-1的赤霉素(GA3), 用量均为100 mL m-2。生长期田间管理同一般高产田。
1.2 测定项目与方法
1.2.1 花生株高测定 花生播种出苗后, 每隔10 d, 用直尺测定子叶节至主茎生长点的长度, 即为花生主茎高。
1.2.2 花生茎秆RNA的提取、纯化、质检和文库构建及其数据分析 花生播种后60 d, 取3个品种及分别喷施外源PP333和GA3的山花108共5组材料的茎秆, 每组材料3次生物学重复, 共计15个样品。剪取植株主茎中部1 cm长度茎秆, 液氮中速冻30 min,-80℃保存, 用于提取茎秆总RNA, 琼脂糖凝胶电泳分析RNA的完整性及是否存在DNA污染; 采用NanoPhotometer分光光度计检测RNA纯度; 采用Qubit 2.0荧光计测量RNA浓度; 经Agilent 2100电泳质检合格后使用, 合格的RNA可用于后续试验。通过Oligo(dT)磁珠与mRNA的ployA尾结合, 富集真核生物的mRNA, 然后将mRNA随机打断。
以片段化的mRNA为模板, 使用六碱基随机引物, 在M-MuLV逆转录酶体系中合成cDNA第1条链。随后用RNaseH降解RNA链, 并在DNA polymerase I体系下, 以dNTPs为原料合成cDNA第2条链。纯化后的双链cDNA经过末端修复、加A尾并连接测序接头。用AMPure XP beads筛选200 bp左右的cDNA, 进行PCR扩增并再次使用AMPure XP beads纯化PCR产物, 最终获得文库。构建完成后, 使用Qubit 2.0荧光计定量。把文库稀释至1.5 ng μL-1后, 使用Agilent 2100生物分析仪对文库的插入片段进行检测。如果插入片段长度符合预期, 再用实时荧光定量PCR对文库有效浓度进行准确定量(文库有效浓度高于2 nmol L-1), 库检合格后用Illumina HiSeq平台进行测序。高通量测序仪得到的图像数据经CASAVA碱基识别转化为序列数据(raw reads)。对原始数据进行去除带接头、poly-N及低质量reads后获得高质量clean reads。并对Q20、Q30、GC含量及重复序列水平进行统计; 将clean reads用HISAT2软件与花生参考基因组(https://peanutbase. org/data/public/Arachis_hypogaea/Tifrunner.gnm1.KYV3/)进行基因比对。
1.3 数据处理与统计
用DPS7.05软件对株高数据进行方差分析和显著性检验, 用Origin2017绘图。通过FPKM方法计算基因的表达量, 随后用Deseq2进行差异基因的筛选。筛选条件为两基因之间的-value < 0.05, Fold change ≤2或≤0.5 (表达上调或下调)。使用R软件语言包clusterProfiler进行差异基因GO功能分析; WGCNA包构建基因共表达网络。
2 结果与分析
2.1 不同品种材料的表型差异与主茎生长动态差异分析
3个品种材料茎高、分枝长差异明显(图1-A)。随播种后天数增加, 各品种及处理的主茎生长均呈S型曲线(图1-B), 播种后10~30 d, 主茎生长缓慢; 40~70 d, 主茎生长迅速, 80~90 d趋于最大主茎高。其中播种后60 d, 山花108与Df216的主茎高均显著低于花育33号, 分别低22.2%和66.3%。与山花108对照相比, 喷施外源GA3可显著提高主茎长, 而喷施多效唑可显著降低主茎高。
2.2 转录组差异表达基因分析
转录组测序数据结果表明, 矮秆Df216与高秆花育33号相比共有5872个差异基因, 其中上调基因2753个, 下调基因3119个; Df216与山花108相比共有6662个差异基因, 其中上调基因2996个, 下调基因3666个; 中间型山花108与花育33号相比共有3832个差异基因, 其中上调基因1673个, 下调基因2159个(图2-A)。3个比较组共有的差异转录基因数目为270个(图2-B)。
PP333: 喷施多效唑的山花108; GA3: 喷施赤霉素的山花108。
PP333: Shanhua 108 sprayed with paclobutrazol; GA3: Shanhua 108 sprayed with gibberellin.
2.3 差异表达基因的GO分类与富集分析
差异基因GO功能注释结果表明, 3个品种两两比较获得GO分类的条目分别有56、54、52个(图3-A); 3组共有51个GO分类的条目, 包含生物过程、分子功能、细胞组成各26、11、14个(图3-B)。3组中, 仅有花育33号 vs. Df216比较组特有1个GO分类条目(GO: 0007610)。以校正后的(adjust)值小于0.05为条件, 筛选显著性的GO条目, 结果表明花育33号 vs. Df216比较组有42个显著富集条目, 特有2个显著富集条目; 山花108 vs. Df216比较组有42个显著富集条目, 特有7个显著富集条目; 山花108 vs.花育33号比较组有31个显著富集条目, 特有6个显著富集条目(图3-C, D; 表1)。
3个比较组共有20个显著富集条目, 主要涉及植物细胞壁及次生细胞壁的生物起源(GO:0042546、GO:0009832和GO:0009834)、细胞壁及次生细胞壁的生物发生调控过程(GO:2000652、GO:1903338)、β-葡聚糖生物合成过程与代谢过程(GO:0051274、GO:0051273)、纤维素生物合成与代谢过程(GO:003 0244、GO:0030243)、氨基酸运输(GO:0006865)等过程(表2)。

表1 不同比较组特有的显著性GO富集
花育33号 vs. Df216与山花108 vs. Df216两比较组共有15个显著富集条目(图3-D), 主要涉及次生细胞壁生物发生(GO:0009833)、苯丙烷生物合成及代谢过程(GO:0009698、GO:0009699)、木质素生物合成过程(GO:0009809)等生物过程; 以及葡萄糖基转移酶活性(GO:0046527)、UDP-糖基转移酶活性(GO:0035251)、纤维素合酶活性(GO:0016759)等分子功能(图4)。
2.4 细胞壁形成相关基因分析
花育33号vs. Df216与山花108 vs. Df216两比较组富集在次级细胞壁生物发生和调控(GO: 0009833和GO:1903338)过程、苯丙烷和木质素合成过程(GO:0009699和GO:0009809)的差异基因主要编码纤维素合酶(cellulose synthase A, CESA)、COBRA-like蛋白、MYB转录因子、苯丙氨酸解氨酶(phenylalanine ammonia-lyase, PAL)、肉桂酸- 4-单加氧酶(cinnamate-4-monooxygenase, CYP 73A)、4-香豆酸辅酶A连接酶(4-coumarate-CoA ligase, 4CL)、肉桂酰辅酶A还原酶(cinnamoyl-CoA reductase, CCR)、莽草酸香豆酯/奎酸酯3’-羟化酶(coumaroyl shikimate/quinate 3’-hydroxylases, C3’H)、咖啡酸3-O-甲基转移酶(caffeic acid 3-O- methyltransferase, COMT)、阿魏酸5-羟化酶(ferulate-5-hydroxylase, F5H)、咖啡酰辅酶A-O-甲基转移酶(caffeoyl-CoA O-methyltransferase, CcoA OMT)等的基因, 与花育33号和山花108相比, 这些基因在Df216中均显著下调(表3)。

表2 不同品种比较组共有的显著性GO富集

(续表2)
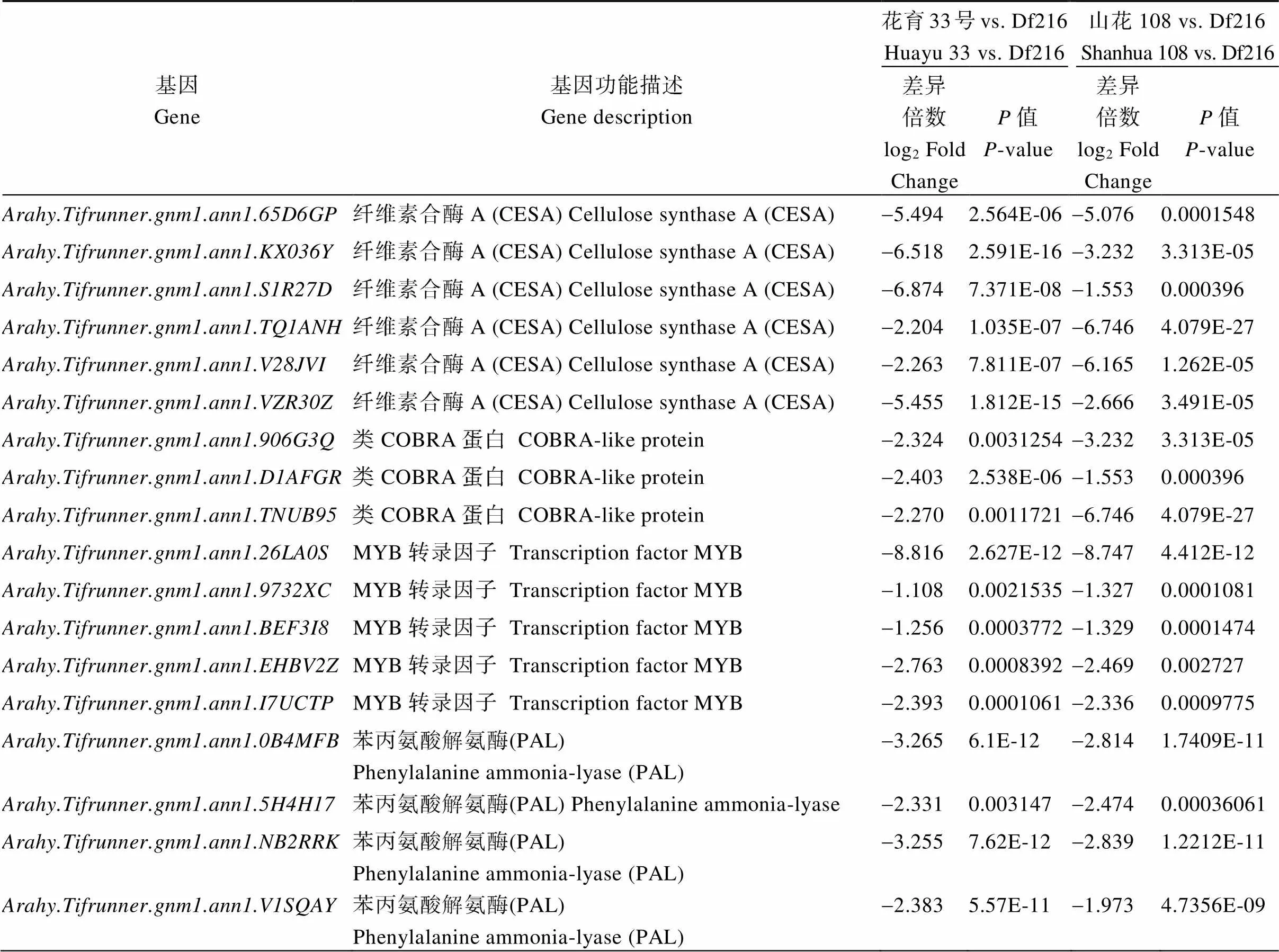
表3 不同比较组细胞壁形成相关基因

(续表3)
2.5 花生基因共表达网络的构建及其与株高相关模块分析
过滤低表达基因后, 共有35,669个基因用于构建加权基因共表达网络。根据基因的表达量进行聚类分析, 检验是否有离群样品, 结果表明, 15个样品聚分为3类, 高秆花育33号为一类、喷施PP333和GA3处理的样品和山花108未喷施处理聚为一类、矮秆Df216为一类(图5-A)。之后利用pickSoftThreshold函数计算选择合适的加权系数β。β的选取标准即满足相关系数的平方接近0.85, 同时还需要保证一定的基因连接度(图5-B)。β值取16来构建共表达网络。
利用blockwiseModules构建网络, 其中最小模块大小(minModuleSize)为30, mergeCutHeight=0.25合并相似性为0.75的模块, 其他参数按照默认设置, 结果如图6-A所示。不同模块用不同颜色表示, 共获得33个模块(Grey模块表示未分配到任何模块的基因), 不同模块间包含基因数差异较大。其中, Turquoise模块包含基因个数最多, 为14,163个; Darkolivegreen模块包含基因个数最少, 为50个(图6-B)。通过共表达网络与株高进行关联, 鉴定到与株高极显著正相关的模块Grey60 (0.79), 其次为Cyan (0.76)和Darkolivegreen (0.7); 极显著负相关的模块Brown (-0.81), 其次为Blue (-0.74) (图6-C)。

表4 模块核心基因及其功能描述
2.6 模块核心基因的挖掘与互作网络分析
选择KME (eigengene connectivity)值最大的基因为每个模块的核心基因, 结果表明Grey60模块核心基因()其功能编码咖啡酰辅酶A-O-甲基转移酶(CCoAOMT); Cyan模块中()基因编码转录因子ATAF2; Brown模块中()基因编码GDSL脂肪酶; Blue模块其核心基因功能尚未鉴定(表4)。
A和B分别表示Grey60和Brown模块。A and B represent Grey60 and Brown modules, respectively.

表5 与模块内核心基因相关联基因及其功能注释

(续表5)
利用Cytoscape软件选取权重(weight)值前10的有关联基因进行可视化, 构建Grey60和Brown模块核心基因的互作网络(图7)。与具有较高互作网络关系的基因分别编码莽草酸香豆酯/奎酸酯3-羟化酶(C3’H)、4-香豆酸辅酶A连接酶(4CL)、羟基肉桂酰辅酶A莽草酸/奎尼酸羟基肉桂酰转移(HCT)、以及快速碱化因子(RALF)、锌指蛋白、类COBRA蛋白等; 与具有较高互作网络关系的基因则分别编码β-1,4-半乳糖基转移酶(GALS3)、果胶乙酰酯酶(PAE)、类受体丝氨酸/苏氨酸蛋白激酶(FEI1)、富含亮氨酸重复序列的伸展蛋白(LRXs)、碱性亮氨酸拉链蛋白(bZIP34)等的基因(表5)。
3 讨论
3.1 茎秆生长相关基因的分析
茎秆的形成是茎端分生组织活动的结果, 茎秆生长受遗传基因调控[21]。20世纪, 以培育矮秆、半矮秆品种为核心的“绿色革命”使小麦、水稻等作物产量获得了大幅提高[22], 一系列矮秆基因得到广泛研究和利用[23-24]。本研究利用转录组测序技术分析3种不同茎高品种茎秆基因表达差异发现, 差异基因主要涉及初生和次生细胞壁的生物起源及调控过程、β-葡聚糖生物合成过程与代谢过程、纤维素生物合成与代谢过程、苯丙烷生物合成及代谢过程、木质素生物合成等过程, 表明这些生物学活动与茎秆生长、株高的形成相关。与禾谷类作物类似, 花生茎秆化学成分主要是纤维素、半纤维素、木质素、果胶质等[25-26]。前人研究发现, 纤维素合酶(CESA)突变体的植株基因表达量降低, 纤维素含量减少, 导致植株矮化[27]。木质素合成相关酶如苯丙氨酸解氨酶(PAL)、肉桂酸-4-单加氧酶(CYP73A)、4-香豆酸辅酶A连接酶(4CL)、肉桂酰辅酶A还原酶(CCR)、莽草酸香豆酯/奎酸酯3’-羟化酶(C3’H)、咖啡酸3-O-甲基转移酶(COMT)、阿魏酸5-羟化酶(F5H)、咖啡酰辅酶A-O-甲基转移酶(CCoAOMT)等突变体或其编码基因表达下调后, 木质素含量降低, 植株表型也呈矮化; 而过表达上述酶后茎明显增高[28-29]。本研究结果表明, 与高秆的花育33号及中间型山花108相比, 矮秆Df216中编码细胞壁CESA基因、上述木质素合成酶等基因均显著下调, 这可能是由于与其编码类COBRA蛋白及转录因子MYB的相关基因也显著下调。研究发现COBRA蛋白是一种胞外GPI锚定蛋白, 其基因表达与CESA基因表达高度相关, 影响细胞壁纤维素合成与细胞伸长, 进而调控茎秆生长影响株高[30-31]。MYB转录因子家族在植物生长发育及代谢调控网络中发挥重要作用[32], 尤其细胞壁中苯丙烷代谢和木质素合成受多种MYB转录因子调控[33]。例如MYB1负向调控木质素合成及细胞壁形成, MYB2则相反[34]。Zhang等[35]研究发现, 超量表达转录因子后, 水稻株高增高。这表明Df216受类COBRA蛋白和转录因子MYB的共同调控, 二者影响CESA及木质素酶基因的表达, 使细胞生长所需纤维素、木质素等合成受阻, 这是导致其茎秆生长缓慢, 株高较低的原因。
3.2 茎秆生长相关共表达模块的鉴定
加权基因共表达网络分析(WGCNA)是一种描述基因关联模式的生物信息学分析方法, 可快速提取出与样品特征相关的基因共表达模块[36]。前人利用WGCNA分析了玉米株高形成等的共表达基因模块[14, 37], 这为挖掘花生茎秆生长调控基因和模块提供了一定参考。本研究利用WGCNA方法对不同茎高花生转录组测序数据构建了加权基因共表达网络,鉴定到与主茎高呈极显著相关的共表达模块: Grey60、Cyan、Darkolivegreen、Brown和Blue。根据模块内基因的连接度, 编码CCoAOMT、转录因子ATAF2、(WAT1)、GDSL脂肪酶的基因分别是Grey60、Cyan、Darkolivegreen、Brown模块的核心基因, 但Blue模块其核心基因功能尚未鉴定到。ATAF2是一种NAC转录因子, NAC在植物衰老、激素应答、胁迫反应等发育过程中起重要作用[38]。直接调控水稻赤霉素合成关键酶如KO与KAO等基因表达, 进而调节水稻茎秆生长[39]。编码一种生长素运载体, 其突变体会导致拟南芥次生细胞壁纤维厚度显著降低[40]。GDSL脂肪酶也参与植物细胞伸长[41]。通过权重值, 本研究构建了Grey60和Brown模块核心基因的互作网络, 基因功能注释显示, 与Grey60核心基因具有较高互作网络关系的基因分别编码C3’H、4CL和HCT, 以及编码快速碱化因子(RALF)、锌指蛋白和类COBRA蛋白等。RALF可通过FERONIA/RIPK受体激酶复合体控制植物细胞的伸长[42]。Huang等[43]的研究发现, 水稻中锌指蛋白OsIDD2可调控木质素合成相关酶CAD等的基因表达, 影响次生细胞壁形成。这表明Grey60模块与细胞壁木质素合成相关, 且受RALF、锌指蛋白和类COBRA蛋白多个因子调控。与Brown核心基因具有较高互作网络关系的基因为编码β-1,4-半乳糖基转移酶(GALS3)、果胶乙酰酯酶(PAE)、类受体丝氨酸/苏氨酸蛋白激酶(FEI1)、富含亮氨酸重复序列的伸展蛋白(LRXs)等基因。GALS3和PAE是果胶合成的重要酶, 其基因的表达可调控次生细胞壁的形成和细胞壁的展延性[44-46]。FEI1和FEI2是富含亮氨酸重复序列类受体类激酶, 介导的信号通常会经相关的转录因子传递到参与细胞壁形成的基因[47]。LRXs参与细胞壁的形成与组装, 超量表达后细胞伸长停止, 导致细胞体积减小, 株高降低[48]。这表明Brown模块与细胞壁果胶合成相关, 且受FEI1和LRXs蛋白介导调控。
4 结论
不同茎高花生茎秆转录组差异基因涉及细胞壁及次生细胞壁纤维素和木质素生物合成过程等生物活动。利用WGCNA方法构建了花生株高的加权共表达网络, 得到极显著关联模块5个, 且鉴定了关联模块内核心基因, 并解析了其基因网络关系, 显示转录因子ATAF2和MYB、快速碱化因子、类COBRA蛋白、锌指蛋白、伸展蛋白、类受体类激酶等介导参与了细胞壁纤维素、木质素和果胶的合成。
[1] Reinhardt D, Kuhlemeier C. Plant architecture., 2002, 3: 846–851.
[2] 张佳蕾, 郭峰, 张凤, 杨莎, 耿耘, 孟静静. 提早化控对高产花生个体发育和群体结构影响. 核农学报, 2018, 32: 2216–2224.
Zhang J L, Guo F, Zhang F, Yang S, Geng Y, Meng J J. Effects of earlier chemical control on ontogeny and population structure of high yield peanut., 2018, 32: 2216–2224 (in Chinese with English abstract).
[3] Flintham J E, Börner A, Worland A J, Gale M D. Optimizing wheat grain yield: effects of(gibberellin-insensitive) dwarfing genes., 1997, 128: 11–25.
[4] Wu J, Kong X Y, Wan J M, Liu X Y, Zhang X, Guo X P, Zhou R H, Zhao G Y, Jing R L, Fu X D, Jia J Z. Dominant and pleiotropic effects of agene in wheat results from a lack of interaction betweenand., 2011, 157: 2120–2130.
[5] Nakamura A, Fujioka S, Sunohara H, Kamiya N, Hong Z, Inukai Y, Miura K, Takatsuto S, Yoshida S, Ueguchi-Tanaka M, Hasegawa Y, Kitano H, Matsuoka M. The role ofand its homologous genes,and, in rice., 2006, 140: 580–590.
[6] Chen W W, Cheng Z J, Liu L L, Man M, You X M, Wang J, Zhang F, Zhou C L, Zhang Z, Zhang H, You S M, Wang Y P, Luo S, Zhang J H, Wang J L, Wang J, Zhao Z C, Guo X P, Lei C L, Zhang X, Lin Q B, Ren Y L, Zhu S S, Wan J M., encoding an HD-Zip II family transcription factor, regulates plant development by modulating gibberellin biosynthesis in rice., 2019, 288: 110208.
[7] Zhang Y X, Yu C S, Lin J Z, Liu J, Liu B, Wang J, Huang A B, Li H Y, Zhao T.regulates plant height and improves grain yield in rice., 2017, 12: e0180825.
[8] Chen X, Lu S C, Wang Y F, Zhang X, Lu B, Luo L Q, Xi D D, Shen J B, Ma H, Ming F. OsNAC2encoding a NAC transcription factor that affects plant height through mediating the gibberellic acid pathway in rice., 2015, 82: 302–314.
[9] Langridge P, Fleury D. Making the most of ‘omics’ for crop breeding., 2011, 29: 33–40.
[10] Van Emon J M. The omics revolution in agricultural research., 2016, 64: 36–44.
[11] Edwards D, Batley J. Plant bioinformatics: from genome to phenome., 2004, 22: 232–237.
[12] Mochida K, Shinozaki K. Genomics and bioinformatics resources for crop improvement., 2010, 51: 497–423.
[13] Wang X D, Zheng M, Liu H F, Zhang L, Chen F, Zhang W, Fan S H, Peng M L, Hu M L, Wang H Z, Zhang J F, Hua W. Fine-mapping and transcriptome analysis of a candidate gene controlling plant height inL., 2020, 13: 42.
[14] 马娟, 曹言勇, 王利锋, 李晶晶, 王浩, 范艳萍, 李会勇. 利用WGCNA鉴定玉米株高和穗位高基因共表达模块. 作物学报, 2020, 46: 385–394.
Ma J, Cao Y Y, Wang L F, Li J J, Wang H, Fan Y P, Li H Y. Identification of gene co-expression modules of maize plant height and ear height by WGCNA., 2020, 46: 385–394 (in Chinese with English abstract).
[15] 巨飞燕, 张思平, 刘绍东, 马慧娟, 陈静, 葛常伟, 沈倩, 张小萌, 刘瑞华, 赵新华, 张永江, 庞朝友. 利用WGCNA进行棉花果枝节间伸长相关基因共表达模块鉴定. 棉花学报, 2019, 31: 403–413.
Ju F Y, Zhang S P, Liu S D, Ma H J, Chen J, Ge C W, Shen Q, Zhang X M, Liu R H, Zhao X H, Zhang Y J, Pang C Y. Identification of co-expression modules of genes related to internode elongation of cotton fruiting branches by WGCNA., 2019, 31: 403–413 (in Chinese with English abstract).
[16] Huang L, Ren X P, Wu B, Li X P, Chen W G, Zhou X J, Chen Y N, Pandey M K, Jiao Y Q, Luo H Y, Lei Y, Varshney R K, Liao B S, Jiang H F. Development and deployment of a high-density linkage map identified quantitative trait loci for plant height in peanut (L.)., 2016, 6: 39478.
[17] Zhang X G, Zhang J H, He X Y, Wang Y, Ma X L, Yin D M. Genome wide association study of major agronomic traits related to domestication in peanut., 2017, 8: 1611.
[18] 彭振英, 单雷, 田海莹, 孟静静, 郭峰, 王兴军, 张智猛, 丁红, 万书波, 李新国. 利用远缘杂交培育半匍匐密枝型高产花生新品系. 中国油料作物学报, 2019, 41: 490–496.
Peng Z Y, Shan L, Tian H Y, Meng J J, Guo F, Wang X J, Zhang Z M, Ding H, Wan S B, Li X G. Breeding of semi-sprawl and dense-branching high yield peanut by distant hybridization., 2019, 41: 490–496 (in Chinese with English abstract).
[19] 鲁清, 刘浩, 李海芬, 陈小平, 洪彦彬, 刘海燕, 李少维, 周桂元, 梁炫强. 花生不同株型主要农艺性状的相关分析及其对单株产量的影响. 热带作物学报, 2019, 40: 1115–1121.
Lu Q, Liu H, Li H F, Chen X P, Hong Y B, Liu H Y, Li S W, Zhou G Y, Liang X Q. Correlation analysis of main agronomic traits of different plant types and path analysis of yield per plant in peanut (L.)., 2019, 40: 1115–1121 (in Chinese with English abstract).
[20] 彭振英, 单雷, 张智猛, 李新国, 万书波. 花生株型与高产. 花生学报, 2019, 48(2): 69–72.
Peng Z Y, Shan L, Zhang Z M, Li X G, Wan S B. High yield and plant type of peanut., 2019, 48(2): 69–72 (in Chinese with English abstract).
[21] 胡珀, 韩天富. 植物茎秆性状形成与发育的分子基础. 植物学通报, 2008, 25: 1–13.
Hu P, Han T F. Molecular basis of stem trait formation and development in plants., 2008, 25: 1–13 (in Chinese with English abstract).
[22] Hedden P. The genes of the green revolution., 2003, 19: 5–9.
[23] Würschum T, Langer S M, Longin C F H, Tucker M R, Leiser W L. A modern green revolution gene for reduced height in wheat., 2017, 92: 892–903.
[24] Asano K, Yamasaki M, Takuno S, Miura K, Katagiri S, Ito T, Doi K, Wu J Z, Ebana K, Matsumoto T, Innan H, Kitano H, Ashikari M, Matsuoka M. Artificial selection for a green revolution gene duringrice domestication., 2011, 108: 11034–11039.
[25] 王健, 朱锦懋, 林青青, 李晓娟, 滕年军, 李振声, 李滨, 张爱民, 林金星. 小麦茎秆结构和细胞壁化学成分对抗压强度的影响. 科学通报, 2006, 51: 679–685.
Wang J, Zhu J M, Lin Q Q, Li X J, Teng N J, Li Z S, Li B, Zhang A M, Lin J X. Effects of stem structure and cell wall chemical composition on resistance to compressive strength in wheat., 2006, 51: 679–685.
[26] 谢星光, 戴传超, 苏春沦, 周家宇, 王宏伟, 王兴祥. 内生真菌对花生残茬腐解及土壤酚酸含量的影响. 生态学报, 2015, 35: 3536–3845.
Xie X G, Dai C C, Su C L, Zhou J Y, Wang H W, Wang X X. Effects of endophytic fungus on decay of peanut residues and phenolic acid concentrations in soil., 2015, 35: 3536–3845 (in Chinese with English abstract).
[27] Xie L Q, Yang C J, Wang X L. Brassinosteroids can regulate cellulose biosynthesis by controlling the expression ofgenes in., 2011, 62: 4495–4506.
[28] Muro-Villanueva F, Mao X Y, Chapple C. Linking phenylpropanoid metabolism, lignin deposition, and plant growth inhibition., 2019, 56: 202–208.
[29] Liu Q Q, Luo L, Zheng L Q. Lignins: biosynthesis and biological functions in plants., 2018, 19: 335.
[30] Yeats T H, Bacic A, Johnson K L. Plant glycosylphatidylinositol anchored proteins at the plasma membrane-cell wall nexus., 2018, 8: 649–669.
[31] Dai X X, You C J, Chen G P, Li X H, Zhang Q F, Wu C Y.encodes a COBRA-like protein that affects cellulose synthesis in rice., 2011, 75: 333–345.
[32] Dubos C, Stracke R, Grotewold E, Weisshaar B, Martin C, Lepiniec L. MYB transcription factors in., 2010, 15: 573–581.
[33] Liu J J, Osbourn A, Ma P D. MYB transcription factors as regualtors of phenylpropanoid metabolism in plants., 2015, 8: 689–708.
[34] Legay S, Sivadon P, Blervacq A S, Pavy N, Baghdady, Tremblay L, Levasseur C, Ladouce N, Lapierre C, Séguin A, Hawkins S, Mackay J, Grima-PettenatiJ., an R2R3 MYB transcription factor from eucalyptus negatively regulates secondary cell wall formation inand poplar., 2010, 188: 774–786.
[35] Zhang Y X, Yu C S, Lin J Z, Liu J, Liu B, Wang J, Huang A B, Li H Y, Zhao T. OsMPH1 regulates plant height and improves grain yield in rice., 2017, 12: e0180825.
[36] Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis., 2008, 9: 559.
[37] Wang H S, Gu L J, Zhang X G, Liu M L, Jiang H Y, Cai R H, Zhao Y, Cheng B J. Global transcriptome and weighted gene co-expression network analyses reveal hybrid-specific modules and candidate genes related to plant height development in maize., 2018, 98: 187–203.
[38] Olsen A N, Ernst H A, Leggio L L, Skriver K. NAC transcription factors: structurally distinct, functionally diverse., 2005, 10: 79–87.
[39] Chen X, Lu S C, Wang Y F, Zhang X, Lv B, Luo L Q, Li D D, Shen J B, Ma H, Ming F.encoding a NAC transcription factor that affects plant height through mediating the gibberellic acid pathway in rice., 2015, 82: 302–314.
[40] Ranocha P, Dima O, Nagy R, Felten J, Corratgé-Faillie C, Novák O, Morreel K, Lacombe B, Martinez Y, Pfrunder S Jin X, Renou J P, Thibaud J B, Ljung K, Fischer U, Martinoia E, Boerjan W, Goffner D.WAT1 is a vacuolar auxin transport facilitator required for auxin homoeostasis., 2013, 4: 2625.
[41] 郝晓云, 蔡永智, 钱雯婕, 袁哈利, 李榕, 李鸿彬. 植物GDSL脂肪酶家族研究进展. 植物生理学报, 2013, 49: 1286–1290.
Hao X Y, Cai Y Z, Qian W J, Yuan H L, Li R, Li H B. Advances in research of GDSL-lipase family in plants., 2013, 49: 1286–1290 (in Chinese with English abstract).
[42] Du C Q, Li X S, Chen J, Chen W J, Li B, Li C Y, Wang L, Li J L, Zhao X Y, Lin J Z, Liu X M, Luan S, Yu F. A Receptor kinase complex transmits RALF peptide signal to inhibit root growth in., 2016, 113: 8326–8334.
[43] Huang P, Yoshida H, Yano K, Kinoshita S, Kawai K, Koketsu E, Hattori M, Takehara S, Huang J, Hirano K, Ordonio R L, Matsuoka M, Ueguchi-Tanaka M. OsIDD2, a zinc finger and INDETERMINATE DOMAIN protein, regulates secondary cell wall formation., 2018, 60: 130–143.
[44] Naran R, Pierce M, Mort A J. Detection and identification of rhamnogalacturonan lyase activity in intercellular spaces of expanding cotton cotyledons., 2007, 50: 95–107.
[45] Liwanag A J M, Ebert B, Verhertbruggen Y, Rennie E A, Rautengarten C, Oikawa A, Andersen M C F, Clausen M H, Scheller H V. Pectin biosynthesis: GALS1 inis a b-1,4-galactan β-1,4-galactosyltransferase., 2012, 24: 5024–5036.
[46] Gou J Y, Miller L M, Hou G C, Yu X H, Chen X Y, Liu C J. Acetylesterase-mediated deacetylation of pectin impairs cell elongation, pollen germination, and plant reproduction., 2012, 24: 50–65.
[47] 张保才, 周奕华. 植物细胞壁形成机制的新进展. 中国科学: 生命科学, 2015, 45: 644–556.
Zhang B C, Zhou Y H. Plant cell wall formation and regulation., 2015, 45: 644–556 (in Chinese with English abstract).
[48] 范春芬, 王艳婷, 彭良才, 丰胜求. 植物细胞壁伸展蛋白的功能与利用. 植物生理学报, 2018, 54: 1279–1287.
Fan C F, Wang Y T, Peng L C, Feng S Q. Plant extensins function and their potential genetic manipulation in crops., 2018, 54: 1279–1287 (in Chinese with English abstract).
Identification of gene co-expression modules of peanut main stem growth by WGCNA
WANG Ying, GAO Fang, LIU Zhao-Xin, ZHAO Ji-Hao, LAI Hua-Jiang, PAN Xiao-Yi, BI Chen, LI Xiang-Dong, and YANG Dong-Qing*
College of Agronomy, Shandong Agricultural University / State Key Laboratory of Crop Biology, Tai’an 271018, Shandong, China
This study was investigated the difference of transcriptome using three different peanut varieties with high main stem by RNA-seq. Transcriptomics combined with weighted gene co-expression network analysis (WGCNA) was used to explore the hub genes related to main stem growth and the molecular mechanisms of morphological formation of peanut stems. Results showed that 5872 differential expressed genes (DEGs) were detected in the Df216 and Huayu 33 comparation group, while 6662 DEGs were detected in the Df216 and Shanhua 108 comparation group. GO analysis suggested that these DEGs were mainly involved in molecular function and biological process, including the primary and secondary cell wall organization and biogenesis, phenylpropanoid biosynthetic and metabolic process, lignin biosynthetic process, and cellulose synthase activity, respectively. There were 33 modules were identified by WGCNA, among which five modules (Grey60, Cyan, Darkolivegreen, Brown, and Blue) were highly significant association with main stem height. According to the connectivity of genes in modules, caffeoyl-CoA O-methyltransferase, transcription factor, WAT1 (), and GDSL esterase/lipasewere the hub genes, respectively. The results of hub gene networks by weighted values indicated that coumaroylquinate 3’-monooxygenase, 4-coumarate-CoA ligase, shikimate O-hydroxycinnamoyltransferase, rapid alkalinization factor,-like protein, and zinc finger protein had high connections within the Grey60 module, while β-1,4-galactosyltransferase, LRR receptor-like serine/threonine-protein kinase, pectin acetylesterase, leucine-rich repeat extensin-like proteinhad high connections within the Brown module. The identification of co-expression modules and their hub genes, and the analysis of gene function and gene networks of key genes will be helpful for revealing the genetic basis of the main height in peanut.
peanut; plant height; transcriptome; weighted gene co-expression network; hub gene
10.3724/SP.J.1006.2021.04223
本研究由国家重点研发计划项目“大田经济作物优质丰产的生理基础与调控” (2018YFD1000900), 山东省重大科技创新工程项目(2018YFJH0601-3), 山东省农业重大应用技术创新项目(SD2019ZZ11)和山东省现代农业产业技术体系花生创新团队首席专家专项基金(SDAIT-04-01)资助。
This study was supported by the National Key Research and Development Program of China “Physiological Basis and Agronomic Management for High-quality and High-yield of Field Cash Crops” (2018YFD1000900), the Shandong Key Research and Development Program (2018YFJH0601-3), the Shandong Agricultural Application Technology Innovation Project (SD2019ZZ11), and the Shandong Modern Agricultural Industrial Technology System Peanut Innovation Team Chief Expert Special Fund (SDAIT-04-01).
杨东清, E-mail: chengyang2364@sdau.edu.cn
E-mail: 15610413063@163.com
2020-10-02;
2021-01-21;
2021-02-20.
URL: https://kns.cnki.net/kcms/detail/11.1809.S.20210220.1422.008.html
