会仙岩溶湿地古菌和细菌群落的共现性
2021-06-05程跃扬靳振江贾远航梁锦桃邱江梅潘复静刘德深
程跃扬, 靳振江, 袁 武, 贾远航, 梁锦桃, 邱江梅,潘复静, 刘德深
(桂林理工大学 a.环境科学与工程学院; b.岩溶地区水污染控制与用水安全保障协同创新中心;c.广西环境污染控制理论与技术重点实验室, 广西 桂林 541006)
0 引 言
土壤微生物参与湿地系统的物质循环, 对维持湿地生态系统具有重要作用[1]。古菌和细菌均为土壤中广泛分布的两大微生物类群, 二者的群落结构均受到多种因素的影响, 其中, 土地利用变化带来植被、管理和土壤理化因子等的变化,都会影响到二者的群落结构, 进而对土壤中的物质循环产生影响[2]。在湿地生态系统中, 古菌、细菌群落以及古菌细菌之间存在复杂的相互作用, 在决定生态系统功能方面起着至关重要的作用。高通量测序技术结合共现网络分析方法能够为研究生态系统内部微生物之间的相互作用提供新的思路。目前, 对湿地微生物之间共现网络的研究主要集中在近岸海洋线虫与细菌和古菌[3]、贝加尔湖细菌与真核微生物[4]以及太湖细菌[5]等方面, 这些研究总体上揭示了不同湿地类型不同微生物群落之间的相互作用, 以及微生物在这些生态系统中生物地球化学循环中的潜在重要作用。
桂林会仙湿地是我国最大的低海拔岩溶湿地[6]。会仙湿地不同土地利用和植物根际细菌群落的丰度和多样性[7-8]及会仙湿地沉积物中甲烷菌的群落结构[9]已有报道, 这些研究表明会仙湿地中细菌和甲烷菌具有较高的多样性。但是, 相比于微生物群落结构和多样性, 探究微生物之间的相互作用对了解生态系统功能更重要, 因此, 需要进一步去探索会仙湿地土壤微生物之间的相互关系。
本文以会仙湿地的湖泊湿地、稻田和稻田撂荒地土壤为研究对象, 以16S rRNA基因的515F和806R为通用引物[10-11], 利用高通量测序方法, 采用共现网络技术来分析细菌群落、古菌群落以及二者之间的共现性, 探究微生物之间的相互作用和共存机制, 以及共存微生物在湿地土壤生物地球化学循环中的重要作用, 更好地理解会仙湿地系统的运行规律, 为会仙湿地的保护提供科学的参考依据。
1 材料与方法
1.1 采样点概况与样品采集
采样点位于广西桂林市西南方向30 km的临桂县会仙镇冯家村, 地处亚热带季风区, 年平均气温18.8 ℃、降雨量1 915.2 mm, 地理坐标为25°01′30″—25°11′15″N, 110°08′15″—110°18′00″E, 采样区域内主要植被有芦苇(Phragmitescommunis)和华科拉莎草(Cladiumchinense)等[12]。近年来, 由于人为垦殖和破坏(很大部分被开发为农业用地, 还有一些是经过长期耕作后的撂荒地), 岩溶湿地现存面积仅24 km2[13], 2018年7月, 在会仙湿地岩溶试验场选取湖泊湿地(优势植被为双穗雀稗草Paspaiumdistichum)、稻田(水稻处于结实期)和稻田撂荒地(优势植被为稗草Echinochloacrusgalli)3种土地利用方式的样地, 在每个样地随机取3个采样点混合为一个样本, 共采集到9个样本。 样本土壤采集深度为0~20 cm, 去除植物根系和碎石等杂物, 一部分自然风干后研磨,分别过 0.85、0.25和0.15 mm筛, 常温存储用于测定理化性质; 另一部分置于-80 ℃冰箱暂时冷藏, 用来提取土壤微生物的DNA。
1.2 土壤养分测定
土壤养分测定方法参照文献[14]进行, 其中土壤pH值的测定采用蒸馏水(无CO2)作为浸提剂, 水土比2.5∶1, 混匀后用pH计(型号: IS128C)测量; 土壤有机碳(SOC)测定采用浓硫酸重铬酸钾外加热法; 土壤全氮(TN)采用浓H2SO4消煮凯氏定氮法; 碱解氮(AN)采用碱解扩散法测定; 总磷(TP)采用碳酸钠熔融法; 速效磷(AP)采用盐酸氟化铵法; 可溶性有机碳(DOC)采用水土振荡提取法。土壤养分含量见表1。

表1 3种土地利用方式土壤的养分含量Table 1 Soil nutrient content in three types of land-use
1.3 土壤DNA提取与高通量测序
用DNA快速提取试剂盒(Power Soil, QIGEN公司)提取土壤的DNA, 提取步骤参照说明书, 使用0.8%琼脂糖凝胶电泳检测DNA。选取引物序列515F(5′-GTG YCA GCM GCC GCG GTA A-3′)和806R(5′-GGA CTA CHV GGG TWT CTA AT-3′)为通用测序引物对古菌和细菌的16S rDNA V4区域进行扩增。PCR(Applied Biosystems® Gene Amp® PCR System 9700)扩增程序包括起始 94 ℃ 、1 min, 然后变性设置为94 ℃、20 s, 退火设置为54 ℃、30 s, 延伸设置为72 ℃、30 s, 并将变性、退火和延伸进行30次循环, 最后为72 ℃ 、5 min。将PCR产物与loading buffer 混合, 使用2%琼脂糖凝胶电泳检测, 胶回收试剂盒QIAquick Gel Extraction Kit (QIAGEN)回收PCR产物, 利用Qubit@2.0 Fluorometer (Thermo Scientific)进行定量, 最后等物质的量混合样本。回收的DNA送到成都罗宁生物科技有限公司, 使用罗宁生物的Hiseq 2500平台PE250模式测序。
得到原始数据后使用Trimmomatic进行质量控制, 再使用Uchime 算法去除嵌合体后得到有效数据。使用UPARSE 算法在97%的一致性对所有OUT进行划分并挑选出代表序列, 使用UCLUST分类法与 SILVA 数据库对OTU进行不同分类水平的注释。利用α多样性指数(包括Chao1、ACE、Shannon、Simpson指数)表征古菌和细菌群落的丰富度和多样性。其中, Chao1和ACE指数用来表征微生物丰富度, Shannon和Simpson指数用来表征微生物群落多样性。
1.4 数据处理
本研究用Excel 2010对实验数据进行初步处理, 用SPSS 24.0软件对α多样性指数、门水平丰度进行方差分析, Duncan法用于不同土地利用方式的显著性检验, 显著性水平为p<0.05, 门水平丰度与环境因子的相关性用Pearson法; 利用 Origin 2017绘制古菌和细菌群落在门水平上的相对丰度图; 使用Cytoscape 3.7.0 软件中的CoNet插件构建古菌细菌共现网络图, 具体操作方法参考顾静馨等[15]提供的步骤, 使用分子复合物检测算法(MCODE)聚类。使用MCODE插件对网络进行模块分析, 所有参数都采用标准值。利用 Network Analyzer 工具, 获取网络的连接度、节点数和连接数。在PASSaGE软件中采用 Mantel分析土壤理化因子对共现网络关键物种的影响。
2 结果与分析
2.1 不同土地利用方式下古菌和细菌多样性
表2列出了3种土地利用方式下的古菌和细菌多样性指数, 可以看出, 稻田撂荒地细菌和古菌的Chao1和ACE指数均低于其他两种土地利用方式, 古菌的Shannon和Simpson指数均为湖泊湿地>稻田>稻田撂荒地; 细菌的Shannon和Simpson指数均为稻田>湖泊湿地>稻田撂荒地, 表明土地利用改变了古菌和细菌的多样性。

表2 3种土地利用方式下的古菌和细菌α多样性指数Table 2 Archaea and bacterial α diversity index in three types of land-use
2.2 不同土地利用方式下古菌和细菌群落特征及与养分含量的相关性
本研究中所得到的古菌序列归属于5个古菌门, 其中, 奇古菌门(Thaumarchaeota)、深古菌门(Bathyarchaeota)和广古菌门(Euryarchaeota)这3个门占总古菌门的99.33%~99.93%(图1a), 奇古菌门和深古菌门的丰度在湖泊湿地、稻田、稻田撂荒地的土壤中差异显著, 分别为68.71%、73.26%、89.71%和23.02%、19.79%、3.67%, Euryarchaeota在3种土地利用方式土壤中的丰度没有显著差异。另外, 本研究所得到的细菌序列归属于39个菌门, 其中, 绿弯菌门(Chloroflexi)、酸杆菌门(Acidobacteria)、变形菌门(Proteobacteria)、厚壁菌门(Firmicutes)、Rokubacteria、硝化螺旋菌门(Nitrospirae)、浮游霉菌(Planctomycetes)、拟杆菌门(Bacteroidetes)、Latescibacteria、芽单胞菌门(Gemmatimatimonadetes)、疣微菌门(Verrucomicrobia)、放线菌门(Actinobacteria)、Epsilonbacteraeota是相对丰度大于1%的细菌门类(图1b), 占细菌总门的96.73%~98.60%。

图1 3种土地利用方式古菌(a)和细菌优势门(b)水平相对丰度Fig.1 Relative abundance of dominant phylun levels of archaea(a) and bacteria(b) in three types of land-use
图2a可以看出, Thaumarchaeota与pH极显著负相关, 与C/N和DOC显著正相关; Bathyarchaeota与pH显著正相关, 与C/N显著负相关, 与DOC极显著负相关; 图2b可以看出, pH与Chloroflexi、Proteobacteria、Nitrospirae和Planctomycetes显著正相关, 与Acidobacteria、Bacteroidetes和Actinobacteria显著负相关; C/N与Rokubacteria、Latescibacteria和Actinobacteria显著正相关。DOC与Acidobacteria、Bacteroidetes和Actinobacteria显著正相关, 与Chloroflexi、Proteobacteria、Nitrospirae显著负相关。
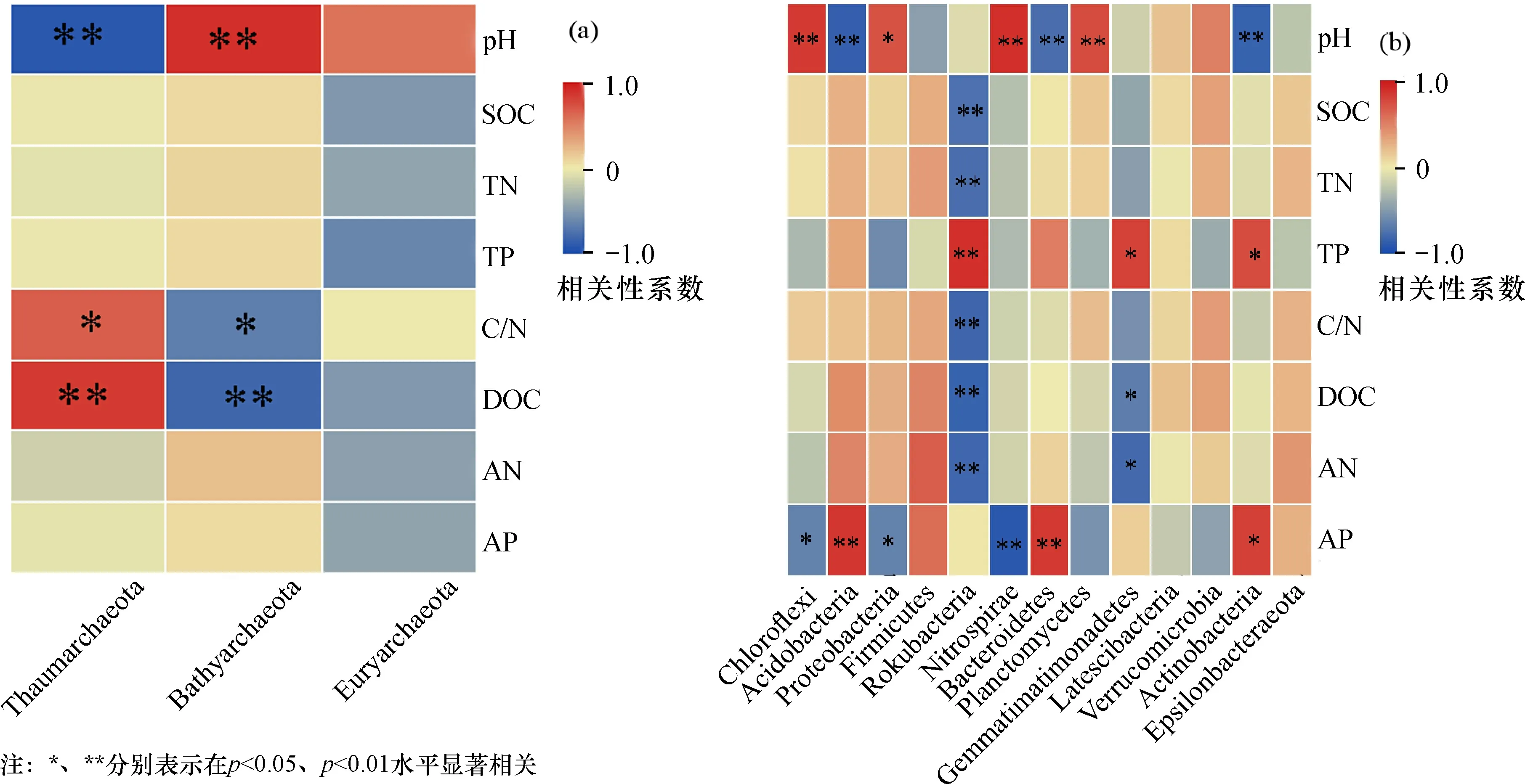
图2 养分因子与古菌、细菌群落之间的相关性热图Fig.2 Heat maps of correlation of nutrient factors with archaea and bacterial communities
2.3 湿地古菌和细菌优势OTUs组成特征
在本研究中, 古菌优势OTUs(相对丰度>0.3%)包括Bathyarchaeota(OTU9、OTU38、OTU85、OTU1093、OTU49、OTU155和OTU231)、Thermoplasmataceae
(热原体科, OTU55、OTU45、OTU227和OTU93)、Ferroplasmaacidarmanusfer1(OTU131)、Methanomassiliicoccales(OTU179)、Methanobacterium(甲烷杆菌属, OTU415)、Methanobrevibactersp. YE315(甲烷杆菌, OTU219)、CandidatusNitrososphaera(OTU240和OTU11)、Nitrosotaleaceae(OTU5)、Nitrososphaeraceae(OTU1、OTU2、OTU2323、OTU204、OTU59和OTU911)。单因素方差分析表明, 排名前10的OUTs中, Nitrososphaeraceae(OTU1和OTU2323)的丰度在3种土地利用方式中有显著差异, 呈现为APF>PF>LW(p<0.05), Nitrososphaeraceae(OTU9、OTU2、OTU38、OTU85、OTU1093)的丰度在PF中显著高于APF(p<0.05), CandidatusNitrososphaera(OTU11)的丰度在LW显著高于PF和APF(p<0.05), Thermoplasmataceae(OTU55)和Nitrososphaeraceae(OTU204)的丰度在3种土地利用中没有显著差异(p<0.05)。
细菌优势OTUs(相对丰度>0.3%)包括Acidobacteria subgroup 6(酸杆菌门GP6, OTU18、OTU68、OTU116、OTU41、OTU58、OTU28、OTU78、OTU180、OTU48、OTU56和OTU345)、Acidobacteria subgroup 4(酸杆菌门GP4, OTU21、OTU613和OTU272)、Holophagae subgroup 7(全噬菌纲GP7, OTU14)、Anaerolineae SBR1031(厌氧绳菌纲, OTU4、OTU12、OTU20、OTU15、OTU22、OTU36)、Anaerolineaceae(厌氧绳菌科, OTU 39、OTU27和OTU52)、Roseiflexaceae(OTU10和OTU30)、Dehalococcoidia(脱卤球菌纲, OTU13)、Chloroflexi(OTU23、OTU66和OTU188)、Helicobactersp. MIT 99-5507(幽门螺杆菌, OTU47)、Ruminococcaceae UCG-002(瘤胃球菌科, OTU67)、Ruminococcaceae UCG-005(OTU230)、Lactobacillusgasseri(格氏乳杆菌, OTU34)、Lactobacillusacidophilus(嗜酸乳杆菌, OTU94)、Lactobacillusmurinus(鼠乳杆菌, OTU69)、Bacteria(OTU54)、Latescibacteria(OTU114)、Leptospirillumsp. Group II ′5-way CG′(OTU8)、Leptospirillumsp. Group II ′C75′(OTU19)、Deltaproteobacteria MBNT15(δ变形菌, OTU24)、AcidiphiliummultivorumAIU301(OTU17)、Neisseriasp. 104(2012)(奈瑟菌, OTU40)、Thauerasp. SWB20(索氏菌, OTU75)、Escherichia-Shigella(大肠杆菌志贺氏菌, OTU185)、Rokubacteria(棒状杆菌门)(OTU44、OTU16、OTU7、OTU25、OTU3和OTU63)、Spirochaetaceae GWE2-31-10(螺旋体科, OTU120)、Candidatus udaeobacter(OTU111)。单因素方差分析表明, 排名前10的OUTs中, Anaerolineae SBR1031(OTU4)、Rokubacteriales(OTU3)和Anaerolineaceae(OTU39)的丰度在LW中显著高于APF(p<0.05), Acidobacteria subgroup 4(OTU21)、Rokubacteriales(OTU16和OTU7)和Acidobacteria subgroup 6(OTU18)的丰度在APF中显著高于LW和PF(p<0.05), Ruminococcaceae UCG-002(OTU67)、Leptospirillumsp. Group II ′5-way CG′(OTU8)和Lactobacillusgasseri(OTU34)在3种土地利用中没有显著差异(p<0.05)。由此可见, 土地利用改变了古菌优势OTUs和细菌优势OTUs的丰度。
2.4 湿地古菌细菌之间、古菌内部和细菌内部和共现网络分析
选择会仙湿地土壤优势古菌与细菌OTUs(相对丰度>0.3%), 用于构建古菌细菌之间群落共现网络图(图3), 然后进一步对古菌内部和细菌内部的网络分别进行模块化分析(图4、图5)。
土壤古菌细菌之间群落的相互作用从图3(不同颜色节点代表古菌或细菌不同的门, 节点上的数值代表古菌或细菌的OTU编号, 节点大小与连接度成正比, 绿色连线表示正相关, 红色连线表示负相关, 下同)可以看出, 会仙湿地土壤中, 古菌细菌共现网络包含了66个显著相关节点, 总的连线有468条(315条为正连线, 153条为负连线), 表明古菌细菌之间群落的相互促进作用大于相互抑制作用。
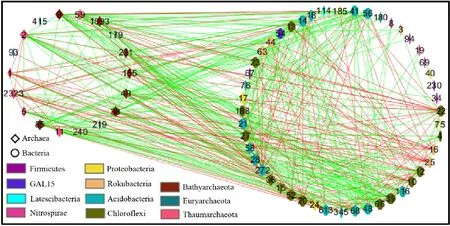
图3 古菌细菌优势OTUs共现网络Fig.3 Co-occurance network of archaea-bacteria dominant OTUs
本研究选取连接度排名前3的OTUs为关键类群, 这些OTUs是OTU9(Bathyarchaeia)、OTU38(Bathyarchaeia)和OTU2(Nitrososphaeraceae)(连接度为27), OTU39(Anaerolineae)(连接度25), OTU85(Bathyarchaeota)、OTU14(Holophagae subgroup 7)和OTU21(Acidobacteria subgroup 4)(连接度24)。为了研究环境因子与共现网络关键物种的相关性, 进行了Mantel test分析(表3), 结果表明, 关键物种与pH极显著正相关, 与C/N和DOC显著正相关。

表3 土壤理化因子与古菌细菌共现网络关键物种的相关性Table 3 Correlation between nutrient factors and key taxa in archaeal-bacteria co-occurrence network
从图4看出, 古菌内部群落的相关性网络由18个节点和37条连线(31条正相关, 6条负相关)构成。在模块1中共同出现了隶属于Bathyarchaeota(OTU85、OTU9、OTU1093、OTU38和OTU155)与Nitrososphaeraceae(OTU2和OTU59)的7个OTUs, 且这些OTUs之间均呈显著正相关关系, 表明它们之间存在相当密切的生态关联。另外OTU2(Nitrososphaeraceae)、OTU38(Bathyarchaeota)、OTU1093(Bathyarchaeota)、OTU9(Bathyarchaeota)、OTU85(Bathyarchaeota)、OTU231(Bathyarchaeota)和OTU155(Bathyarchaeota)是古菌关键类群。MCODE分析古菌相关性网络得到模块的网络分数分别为6.00和3.33, 并且关键类群均出现在了分数高的模块1中。
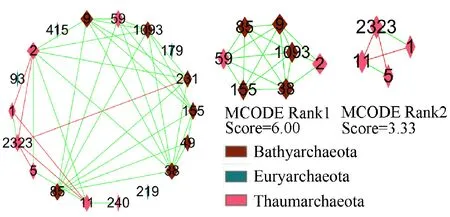
图4 古菌优势OTUs共现网络及模块Fig.4 Co-occurance network and module of archaea dominant OTUs
从图5看出, 细菌共现网络由45个节点和227条连线组成(149条正相关, 78条负相关)构成。从细菌内部网络的模块1可以看出OTU613 、OTU21和OTU272(Acidobacteria GP4)与OTU52、OTU12、OTU27、OTU66、OTU13、OTU23和OTU188(Chloroflexi)呈显著的负相关关系, 说明这两类菌可能存在竞争关系。此外, 除了OTU613、OTU21和OTU272外, 模块1中的其它微生物之间均呈现显著的正相关关系, 说明这些微生物可能存在共存关系。而细菌内部共现网络的关键类群是OTU21(Pyrinomonadaceae RB41)、OTU48(Acidobacteria subgroup 6)和OTU44(Rokubacteria)。MCODE分析古菌相关性网络得到的子网络的网络分数分别为10.76和3.00; 模块1中的互作关系最为紧密, 且关键类群均出现在模块1中。

图5 优势细菌OTUs共现网络图及模块Fig.5 Co-occurance network and module of bacteria dominant OTUs
3 讨 论
3.1 土地利用方式对古菌和细菌群落结构和多样性的影响
有研究发现古菌多样性在稻田土壤显著高于稻田撂荒地(表2), 养分含量与古菌数量和多样性呈显著正相关关系[16-17]。 在本研究中, 与稻田撂荒地相比, 稻田施肥提高了碳、氮和磷等营养物质, 促进了古菌的生长和代谢, 古菌多样性提高。3种土地利用方式的优势古菌均为Thaumarchaeota、Bathyarchaeota和Euryarchaeota, 表明湿地系统的微生物群落在门水平上具有一定的稳定性, 这样有利于保证湿地系统服务功能的相对稳定。Thaumarchaeota和Bathyarchaeota的古菌相对丰度在不同土地利用类型之间存在差异。稻田撂荒地的最优势菌门是Thaumarchaeota, 该门中检测出的微生物均为氨氧化古菌(Candidatusnitrososphaera、Candidatusnitrosotenuis), 这是因为本研究设计的撂荒地的pH值较低, 大量氨分子转化为铵根离子, 而氨氧化古菌长期适应低氨环境, 高效地把环境中较低浓度的氨分子氧化[18]。Thaumarchaeota喜好低氮和高碳环境[19], 稻田撂荒地的碳氮比和DOC较高, 为该菌提供了适宜的生长环境, 所以稻田撂荒地的Thaumarchaeota丰度较高; 另外, 深古菌门的丰度也占了较大比例, 这与吴麒等[9]对会仙湿地深古菌的丰度较高的研究结果一致。有研究发现, 该门的古菌能利用二氧化碳和氢气作为能源物质进行自养合成乙酸[20], 同时还能降解芳香烃和纤维素等有机化合物进行异养代谢产生乙酸, 由此推测该古菌群落属于既能自养又能异养的产乙酸菌[21], 因此在土壤有机质的转化中具有重要的作用。该菌为厌氧微生物群落的主要组成部分[22], 湖泊湿地和稻田的厌氧条件为此类菌提供适宜的生存环境, 以至于深古菌丰度高于稻田撂荒地。此外, Euryarchaeota的丰度在3种土地利用方式下没有显著差异, 且该菌门水平下包含了大量厌氧产甲烷菌, 厌氧产甲烷菌属于Euryarchaeota, 主要存在湿地、海洋、湖泊沉积物和动物粪便等环境中[23-25]。而本研究区域附近村民散养的鸭子活动较多, 带入的粪便可能对该菌丰度提高有促进作用。
稻田的细菌多样性比湖泊湿地和稻田撂荒地高, 与贾远航等[7]对会仙湿地的研究结果基本一致, 表明湿地土壤经过多年的稻作以后, 稻田土壤有机碳和总氮量逐渐提高, 促进了细菌多样性的提高, 而撂荒以后稻田土壤养分下降, 细菌多样性降低。进一步分析发现, 3种土地利用方式的优势菌在门水平上均为Chloroflexi、Acidobacteria和Proteobacteria。这与文献[26]报道的会仙湿地细菌群落优势门的研究结果一致, 也与来自全国42个湿地土壤大样本细菌的研究结果一致[27], 说明这3个门的细菌群落对湿地环境的高度适应性, 在湿地土壤生物地球化学循环中具有潜在的重要作用, 从而可能维持了湿地土壤生态系统的稳定。
3.2 湿地古菌细菌、古菌和细菌的共现网络分析
在自然界中, 微生物群落形成了复杂的生态网络, 基于生态网络, 可以推测物种间的相互作用。处于互利共生、共栖和共聚集等的微生物呈正相关关系, 而处于竞争、偏害共生和捕食等关系的微生物为负相关[28]。图2表明, 会仙湿地的古菌细菌共现网络中的正相关连线多于负相关连线, 其原因有: ①在长期的进化过程中, 二者对氧的需求条件不同。 细菌大多属于好氧菌或兼性厌氧菌, 而古菌大多为厌氧菌。二者占据不同的生态位, 生态位之间的互补关系有利于整个湿地微生物群落之间的共生。如绿弯菌等类群是需氧或兼性厌氧菌, 它们在分解有机物的过程中消耗了大量的氧气[29], 降低了土壤中的氧分压, 从而有利于深古菌等在厌氧环境中占优势的古菌的生存[30]。②不同微生物对有机化合物(或无机物)的利用不同, 如深古菌和变形菌等可以把高分子化合物降解为低分子的有机物, 这些低分子量的有机物为酸杆菌等的生长提供了可利用的碳源[21-31]。因此, 这些微生物群落之间的协同作用, 共同维护了湿地生态系统微生物群落的多样性, 在湿地土壤的生物地球化学循环过程中协同发挥作用, 进而维持了湿地生态系统的功能稳定。碳和氮等能源物质的减少会导致微生物之间的竞争关系大于协同关系[32-33], 说明能源物质丰富的环境有利于微生物的繁殖和物质交换, 进而减少微生物之间的负相互作用[34]。关键类群是微生物群落里具有高度连接的物种, 这些物种的增加或减少都会引起整个生态网络相互作用的变化[35-36]。本文把连接度排名前3的OTUs定义为关键类群, 通过Mantel分析发现, 影响这些关键类群的因子是pH、C/N和DOC, 这与古菌和细菌优势门的影响因子一样, 说明这3个环境因子不仅影响古菌和细菌的群落结构, 还影响古菌细菌的互作关系及生态网络的稳定。
进一步对古菌细菌共现网络里的古菌内部网络(图3)研究发现, 古菌群落也是相互促进作用大于相互抑制作用。深古菌门与奇古菌门在节点、连接度和模块中占主导地位, 说明这两个门的古菌在会仙湿地古菌所参与的生物地球化学循环中可能具有潜在的重要作用。此外, 在模块1中共同出现了分属Bathyarchaeota与Nitrososphaeraceae的OTUs, 且这些OTUs之间均呈显著的正相关关系, 表明它们之间相互合作来共同适应湿地环境。在Nitrososphaeraceae中, 草酰乙酸可促进Nitrosotalea的生长, 低浓度的丙酮酸也能够促进NitrososphaeraviennensisEN76的快速生长,说明Nitrososphaeraceae中不同的微生物对不同有机物的响应不同[37]。 尽管3种土地利用方式下土壤pH、总有机碳和溶解性有机碳在不同程度上有所差异, 但这些小分子有机物与氨氧化古菌之间的关系值得进一步研究。隶属于深古菌门的微生物能把高分子有机化合物芳香烃和纤维素降解为乙酸[21], 这为氨氧化古菌提供了可利用的碳源, 促进了氨氧化古菌群落生长, 因此Nitrososphaeraceae的丰度提高。
从细菌内部网络的模块1(图4)可以看出, Acidobacteria GP4与Chloroflexi之间为显著的负相关关系, 说明这两类细菌可能生态位相似, 因而存在竞争关系。 相似的是, Acidobacteria GP4与Chloroflexi呈显著的负相关关系也出现在玉米轮作的土壤中[38], 这是因为Acidobacteria GP4等通过产生抗菌剂或增强了植物内生细菌对植物病原体的拮抗作用成为益生菌, Chloroflexi不能固定氮, 与植物争夺氮资源成为病原菌。而植物的发病机制是病原菌和益生菌相互竞争的过程[39], 因此, 二者之间的竞争关系可能在一定程度上决定着会仙岩溶湿地植被的健康程度。此外, 除OTU613 、OTU21和OTU272外, 模块1中的其他微生物之间均为正相关关系, 如Proteobacteria(OTU17)与Acidobacteria GP6(OTU48、OTU58和OTU56)呈显著正相关关系。Proteobacteria的细菌在有机物分解过程中发挥着关键作用, 该类菌能利用纤维素酶、几丁质酶和淀粉酶等产生大量低聚糖和芳香醇, 而这些低聚糖和芳香醇可以被Acidobacteria的细菌作为碳源[31], 表明Proteobacteria与Acidobacteria GP6这两类微生物可能协同作用, 促进了土壤中大分子有机物的降解。
3.3 湿地系统关键微生物及其对模块化的影响
古菌内部群落的关键类群是隶属于Bathyarchaeota和Nitrososphaeraceae的7个OTUs, 细菌内部群落共现网络关键类群隶属于Pyrinomonadaceae、Acidobacteria subgroup 6和Methylomirabilaceae的3个OTUs。这些关键类群对于维持会仙湿地微生物生态网络的稳定具有重要作用。另外, 本文研究发现古菌内部和细菌内部的关键类群均出现在各自得分较高的模块中, 这与马泊泊等[40]的关键类群出现在得分较高的模块中的结论一致。这是因为微生物的聚集是以功能决定的, 关键类群在决定微生物群落功能上也有重要作用, 因此功能微生物之间共存网络值得进一步研究。
4 结 论
(1)会仙湿地系统土壤的优势古菌门为Thaumarchaeota、Bathyarchaeota和Euryarchaeota, 但Thaumarchaeota在稻田撂荒地丰度较高, Bathyarchaeota在湖泊湿地丰度较高。另外, 湿地系统土壤的优势细菌门为Chloroflexi、Acidobacteria和Proteobacteria。影响古菌群落的主要环境因子是pH、C/N和DOC。
(2)关键的古菌隶属于Bathyarchaeota和Nitrososphaeraceae, 关键的细菌隶属于Pyrinomonadaceae、Acidobacteria subgroup 6和Rokubacteria, 影响古菌细菌之间相互作用的关键微生物隶属于Bathyarchaeota、Nitrososphaeraceae、Anaerolineaceae、Holophagae Subgroup 7和Pyrinomonadaceae。
(3)古菌细菌、古菌和细菌的共现网络均表明古菌细菌、古菌和细菌的相互促进作用大于相互抑制作用。影响古菌细菌共现网络关键类群的主要环境因子是pH、C/N和DOC。
