(2R,3R)-1,4-二甲氧基-1,1,4,4-四苯基-2,3-丁二醇合成方法研究进展
2021-06-03胡晓允单自兴
胡晓允,单自兴
1中南民族大学化学与材料科学学院,武汉 430074
2武汉大学化学与分子科学学院,武汉 430072
(2R,3R)-1,4-二甲氧基-1,1,4,4-四苯基-2,3-丁二醇(1)是天然酒石酸衍生的一种 C2-对称的位阻型手性二醇,在有机合成中有着广泛的应用。(2R,3R)-1作为手性助剂在硼酸手性化学中的研究最为深入,如作为手性硼酸的保护基团用于不对称环丙烷化、环氧化、3,3-Sigmatropic重排和羰基的烯丙基化反应。2012年,Pietruszka等[1]就(2R,3R)-1的制备和应用专门撰写了一篇综述。
尽管(2R,3R)-1已经发展为一类应用广泛的手性二醇,但其制备方法仍停留在30年前的合成方案,关于它的合成研究报道较少。最早报道(2R,3R)-1的合成方案是从天然酒石酸酸酯出发,经5步反应制得,其关键步骤是光学纯酒石酸酯仲羟基的保护及去保护和叔羟基的甲基化程序:仲羟基的去保护涉及到2,3-二氯-5,6-二氰基苯醌(DDQ)氧化和LiAlH4还原程序,而叔羟基的甲基化则需要利用NaH/MeI等试剂[2]。2009年开始,本课题组对(2R,3R)-1,1,4,4-四取代丁四醇的制备及区域选择性转变化学进行了持续的研究,基于(2R,3R)-1,1,4,4-四苯基丁四醇(2)的区域选择性 2,3-螺硼化反应(硼化学保护)和 2,3-亚硫酸酯化反应(环亚硫酸酯保护),相继发展了两种方案制备(2R,3R)-1。前者避免了DDQ-LiAlH4氧化-还原去保护程序,但仍采用NaH/MeI体系进行甲基化,且利用HF进行水解脱硼,在制备成本和环境保护方面存在一些问题。环亚硫酸酯保护方案利用常规试剂甲醇引入甲氧基,避免了NaH/MeI试剂的使用;另一方面利用无机碱液进行去保护,取代了DDQ-LiAlH4氧化-还原去保护程序。相对于原合成方案,本课题组发展的合成方案缩短了合成路线,降低了制备成本,具有高效、便捷、绿色化程度较高的优势。本文主要对(2R,3R)-1合成方法的研究进展进行概述。
1 缩醛保护法制备(2R,3R)-1
1.1 Nakayama合成路线
(2R,3R)-1的合成路线最初是由Nakayama和Rainier[2]在1990年提出来。如图1所示,天然酒石酸甲酯与对甲氧基苯甲醛的缩醛化合物在对甲苯磺酸催化下发生醇置换,生成的缩醛将酒石酸酯2,3-位仲羟基保护起来,继而与苯基格氏试剂反应,对酯基进行烃基化生成手性二醇。接下来,利用NaH/MeI对新生成的两个叔羟基进行醚化。最后,用DDQ进行氧化,得到羟基酯,再用LiAlH4还原将两个仲羟基游离出来,通过5步反应以37%的总收率得到(2R,3R)-1。
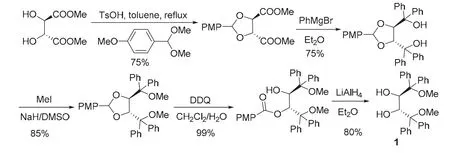
图1 Nakayama合成路线[2]
缩醛在酸性条件下易水解得到相应的醛和醇。但 Nakayama合成方案中,形成的缩醛稳定性较好,且酸性条件下,新生成的甲氧基易被水解。所以采用DDQ-LiAlH4进行氧化-还原将2,3-位仲羟基游离出来。这无疑增加了制备(2R,3R)-1的成本和合成操作的繁琐,尤其 LiAlH4的使用需要无水操作,大量使用具有一定的危险性。
1.2 Nakayama合成路线的改进
2008年,Pietruszka研究组[3]对该合成方案进行了改进。如图2所示,采用便宜的无机盐溴酸钠和连二亚硫酸钠代替DDQ对缩醛进行氧化,但仍需用LiAlH4进行还原,其他步骤和Nakayama方案一致。出乎意料的是,该合成方案中,利用苯基格氏试剂对酯基进行烃基化时,Pietruszka采用甲基四氢呋喃代替乙醚做溶剂,产率由75%提高到98%,反应总收率提高到48%。
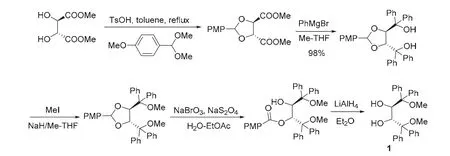
图2 Pietruszka研究组合成路线[3]
2 (2R,3R)-2的区域选择性转变制备(2R,3R)-1
(2R,3R)-2 是 TADDOL (α,α,α’,α’-tetraaryl-2,2-dimethyl-1,3-dioxolan-4,5-dimethanol)母体化合物,尽管TADDOL是一种通用的手性二醇,在不对称合成中应用广泛[4,5],但关于其母体化合物(2R,3R)-2的研究却很少。2010年,本课题组尝试通过硼化学方法将酒石酸酯的仲羟基保护起来,如图3所示,最初设计合成路线Route A:通过苯基硼酸将酒石酸二乙酯2,3-仲羟基保护起来,继而与苯基格氏试剂反应,再水解脱去硼保护基得到(2R,3R)-2。考虑到苯基硼酸一般通过硼酸酯与苯基格氏试剂制备,其成本相对比较高昂,又设计合成路线Route B:硼酸三丁酯代替苯基硼酸对酒石酸酯的仲羟基进行保护。这两种方案制备(2R,3R)-2的产率基本相当,但第二条路线无疑成本要低廉些。上面两条合成路线相对于传统的缩醛保护和去保护方法成本低廉、操作方便,但还需要保护和去保护程序,且浪费了硼试剂。为了进一步简化实验步骤,提高原子经济性,我们设计合成路线Route C:苯基格氏试剂直接与酒石酸二乙酯“一锅”反应制备(2R,3R)-2。尽管该合成方案浪费了格氏试剂,但缩短了反应的操作程序,这三条合成路线以基本相当的产率得到目标化合物。因此,我们选定酒石酸酯与格氏试剂“一锅”法合成了一系列(2R,3R)-1,1,4,4-四取代丁四醇[6]。
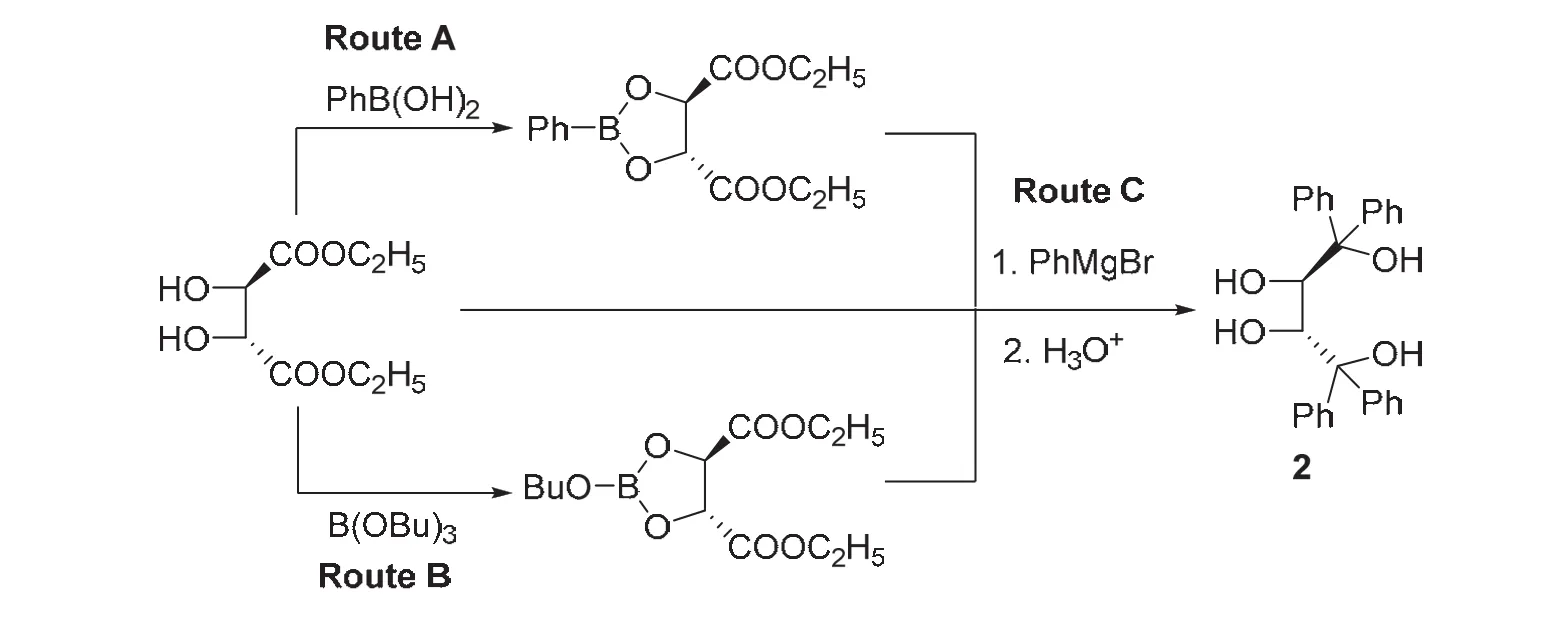
图3 (2R,3R)-2的合成路线[6]
2.1 (2R,3R)-2硼化学保护法合成(2R,3R)-1
(2R,3R)-2有两个仲羟基和两个叔羟基,叔羟基所连碳原子上有苯基取代基,空间位阻和电子性质决定仲羟基和叔羟基应具有不同的反应行为。我们设想利用仲羟基和叔羟基反应活性的差异及苯基取代基的空间位阻对其实现选择性衍生化,避开缩醛保护和去保护的繁琐步骤。经过探索,我们发现(2R,3R)-2与不同的硼试剂反应时,会表现出不同的区域选择性。如图4所示,(2R,3R)-2与烃基硼酸反应时,会发生高度区域选择性 1,3-环硼化反应,生成手性双环[4.4.0]二硼酸酯(3)[7];但在碱性条件下,与硼酸则发生高度区域选择性2,3-螺硼化反应生成手性螺硼酸盐(4)[8]。
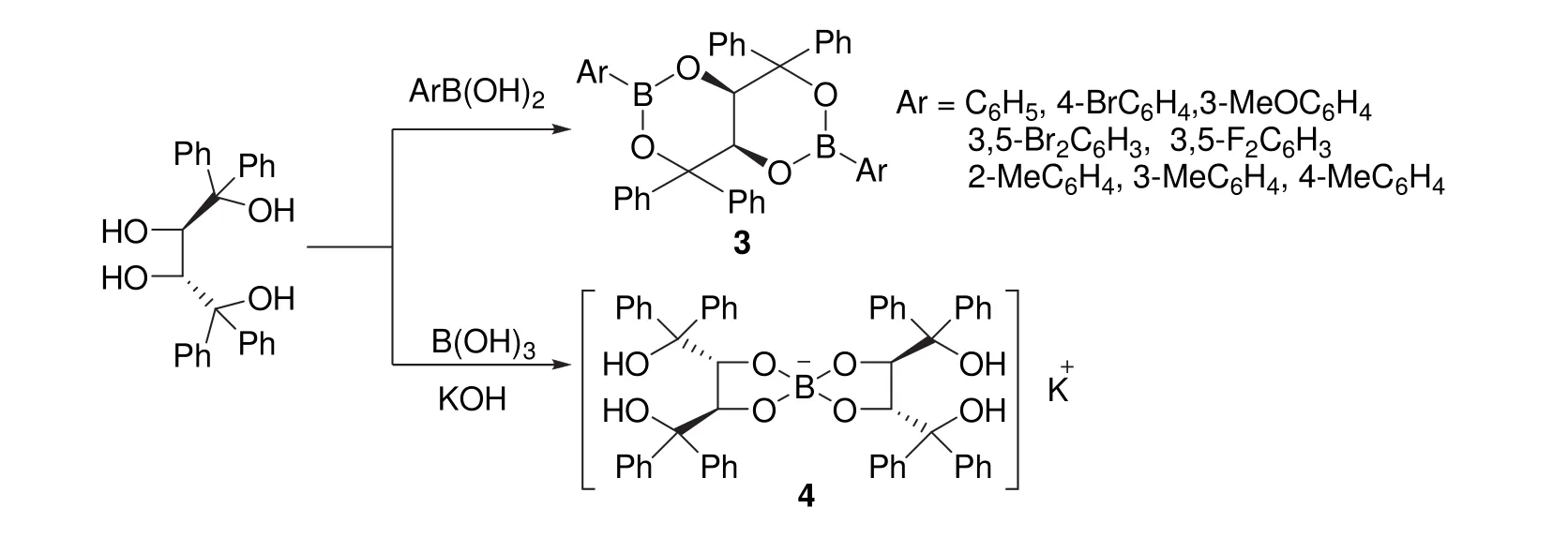
图4 (2R,3R)-2与硼试剂的区域选择性衍生化[7,8]
手性螺硼酸盐4性质很稳定,且容易制备,通过一个硼原子将两分子(2R,3R)-2的仲羟基选择性地保护起来。利用这一特性,我们设计如下合成路线制备(2R,3R)-1。如图5所示,天然酒石酸二乙酯与苯基格氏试剂“一锅”反应制得(2R,3R)-2,在强碱KOH溶液存在下,(2R,3R)-2与硼酸发生高度区域选择性 2,3-螺硼化反应,近乎定量地得到手性螺硼酸钾盐 4。接下来,通过传统的 NaH/MeI甲基化程序对手性螺硼酸盐4的叔羟基进行醚化,最后用HF的甲醇溶液进行去保护,将两个仲羟基游离出来,高效地制得(2R,3R)-1。该合成方案用廉价的硼酸和无机酸碱取代了DDQ和LiAlH4等试剂,简化了合成方案,将报道的5步合成反应优化至4步,反应总收率达到45%[8]。
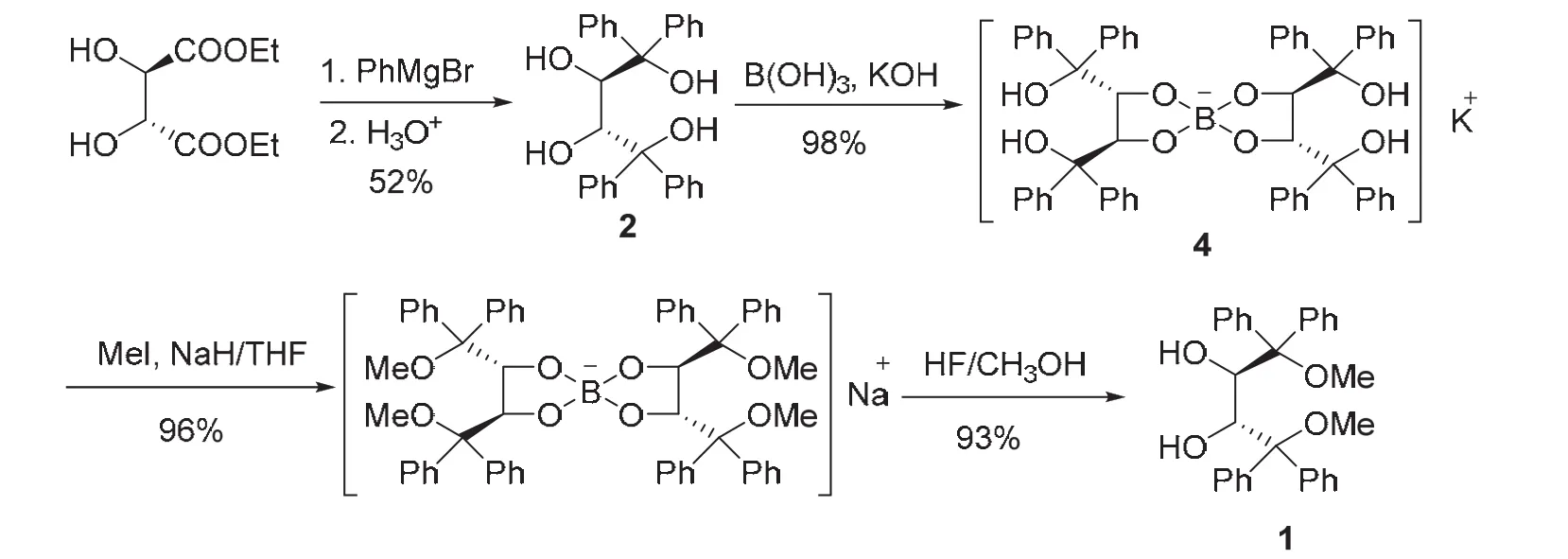
图5 硼化学保护法合成路线[8]
尽管该合成方案提高了原子利用率,简化了合成方案,但仍然存在以下两点不足:1) 1,4-位甲氧基的引入采用传统的醚化试剂MeI/NaH,制备成本仍然较高,工业生产一般用硫酸二甲酯做甲基化试剂,但硫酸二甲酯和MeI均为毒性较大的试剂;2) 反应最后切断B―O键需要使用HF。HF不仅毒性较大,还具有极强的腐蚀性,能强烈地腐蚀金属、玻璃和含硅的物体,一般需要在塑料容器中开展实验。所以从绿色化学的角度审视该合成方案,仍有继续优化的空间。
2.2 (2R,3R)-2环亚硫酸酯保护法合成(2R,3R)-1
我们在考查(2R,3R)-2的反应化学时,发现在一定条件下,其能与氯化亚砜发生高度区域选择性2,3-环亚硫酸酯化反应,生成双羟基环亚硫酸酯(4R,5R)-5,游离的叔羟基与氯化亚砜进一步反应可以被氯代,得到二氯代环亚硫酸酯(4R,5R)-6。研究发现,控制氯化亚砜的用量,也可以实现(2R,3R)-2与氯化亚砜一步反应直接制得(4R,5R)-6 (图6)[9]。
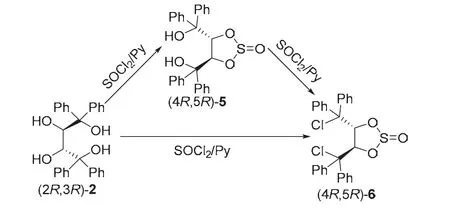
图6 (2R,3R)-2与氯化亚砜的区域选择性环亚硫酸酯化反应[9]
我们知道,卤代烃在有机合成中是关键的合成中间体,亲核取代反应是其典型的化学性质之一,C―Cl极性键易于被各种亲核试剂进攻,引入相应的官能团。我们设想利用甲醇作为亲核试剂与二氯代环亚硫酸酯(4R,5R)-6反应,引入甲氧基,从而避开羟基的甲基化程序。基于这一设想,设计了如下合成方案,如图7所示,天然酒石酸酯二乙酯经过烃基化反应制得(2R,3R)-2,与氯化亚砜发生高度区域选择性2,3-环亚硫酸酯化反应生成(4R,5R)-6,与甲醇发生亲核取代反应引入甲氧基,形成双甲氧基环亚硫酸酯(4R,5R)-7,用碱液处理脱去亚硫酰基,经4步反应,以45%的总收率制得(2R,3R)-1[9]。
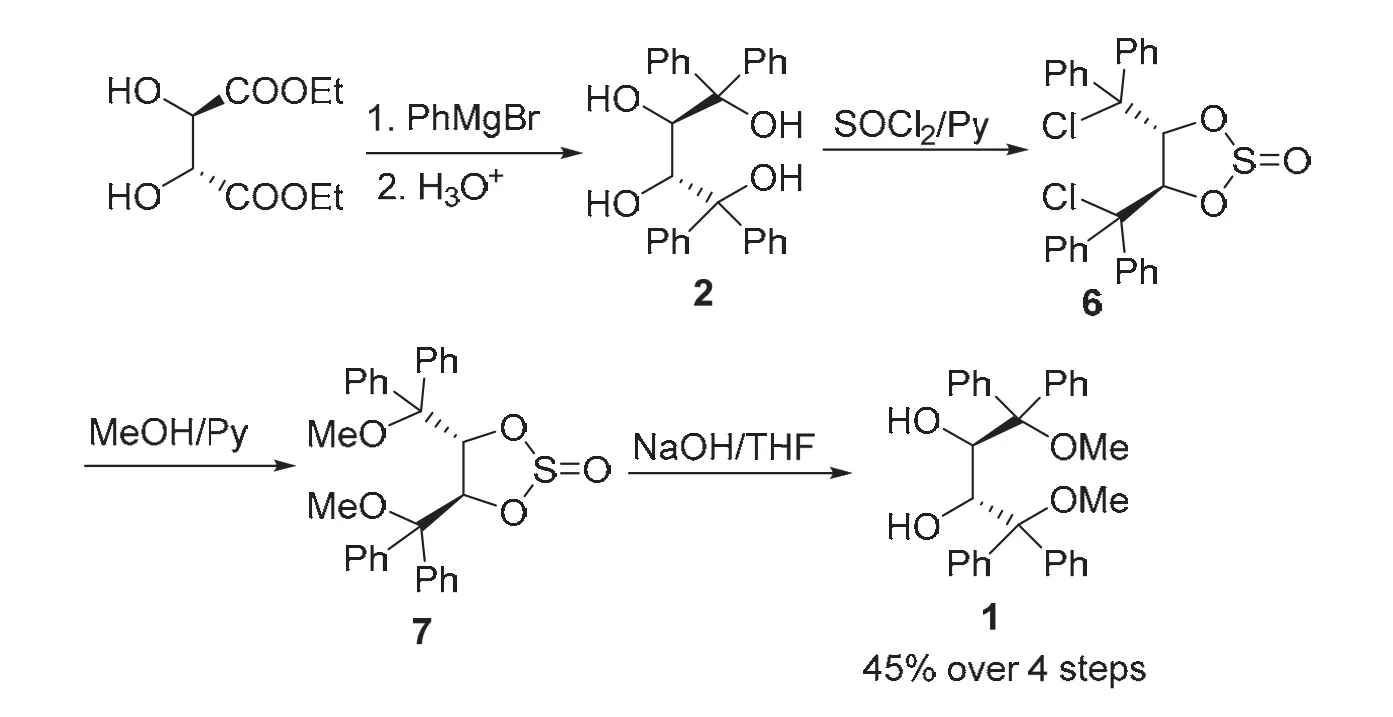
图7 环亚硫酸酯保护法合成路线[9]
该合成方案利用亚硫酰基实现对酒石酸酯仲羟基的保护,而其去保护通过常规碱液即可实现,相对于缩醛和螺硼酸盐的去保护方法,该方法高效便捷,易于大规模制备;另一方面,利用甲醇与卤代烃的亲核取代反应引入甲氧基,避开了通过MeI/NaH对羟基醚化的方案,不仅大大降低了制备成本,简化了实验操作步骤,而且避免了毒性较大的甲基化试剂的使用,提高了该合成方案的绿色化程度。
旋光测定揭示在通过(2R,3R)-2的区域选择性2,3-环亚硫酸酯化制备(2R,3R)-1过程中,即使用强碱氢氧化钠溶液处理,手性中心也未发生消旋化。由此推测在环亚硫酸酯水解过程中,断键反应未涉及到手性中心。基于该实验结果,提出环亚硫酸酯(4R,5R)-7水解机理如图8所示:OH−首先进攻环亚硫酸酯的硫原子,而非手性碳原子。亚硫酰基断键,接下来S-O键断裂,新生成的O−夺取水中的H质子形成羟基,OH−继续进攻硫原子,再断去另一个S-O键,从而游离出两个仲羟基[9]。
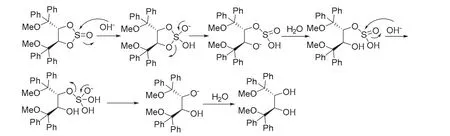
图8 环亚硫酸酯(4R,5R)-7可能的水解机理[9]
3 结语
(2R,3R)-1合成方法的不断优化,是从绿色化学的角度去审视传统的合成方案,通过高度区域选择性控制,发展出高效、便捷、绿色化程度较高的合成方案。以绿色化学理念审视已有的合成方案,开发具有高度选择性,包括化学选择性、区域选择性及立体选择性的合成路线,提高合成方案的原子经济性,这在有机合成化学中仍极具发展空间。
