美国生物药上市后风险管理及对我国的启示
2021-05-10李梦颖王峻霞蒋蓉
李梦颖 王峻霞 蒋蓉
摘 要 目的:借鉴美国生物药上市后风险管理经验,为我国生物药上市后风险管理提供参考。方法:通过研究美国FDA发布的指南文件、网站信息等资料,对美国生物药上市后风险管理进行分析,并以英夫利昔单抗为例介绍其具体实施情况,总结其管理特点并提出对我国生物药上市后风险管理的启示及相关建议。结果与结论:美国生物药上市后的风险管理主要包括“风险评估与降低策略(REMS)”“上市后研究和臨床试验制度”两方面,其中后者包括上市后要求(PMR)和上市后承诺(PMC)两类。以英夫利昔单抗为例,该药自1998年8月被美国FDA批准上市后,其生产厂家于2009年向FDA提交了REMS并获批,并先后5次提出上市后研究和临床试验。可知美国生物药上市后风险管理是由FDA通过出台具体指南,鼓励多角色参与风险管理,实现与患者的有效沟通,对生物药风险进行持续监管,以降低生物药的使用风险。对于生物药而言,我国尚未制定系统、具体的实施细则和指南,在上市后风险管理方面仍存在欠缺。建议我国可借鉴美国对生物药上市后风险管理的措施和制度,尽可能吸纳利益相关方参与上市后管理,通过与患者进行有效沟通以提升患者用药风险意识,并且进一步完善上市后研究管理制度,保障患者用药安全。
关键词 美国;生物药;上市后风险管理;启示
中图分类号 R951;S859.79+7 文献标志码 A 文章编号 1001-0408(2021)07-0776-06
ABSTRACT OBJECTIVE: To learn from the experience of post-marketing risk management of biopharmaceuticals in the United States, and to provide reference for post-marketing risk management of biopharmaceuticals in China. METHODS: By studying guidance documents and website information issued by FDA, the risk management of biopharmaceuticals after marketing in the United States was analyzed. Taking infliximab as an example, the specific implementation situation was introduced, the management characteristics were summarized, and the enlightenment and relevant suggestions were put forward for the risk management of biopharmaceuticals after marketing in China. RESULTS & CONCLUSIONS: The post-marketing risk management of biopharmaceuticals in the United States mainly includes two aspects as “risk evaluation and mitigation strategy (REMS)” and “post-marketing study and clinical trials system”. The latter included post-marketing requirement (PMR) and post-marketing commitment (PMC). Taking infliximab as an example, since it was approved by FDA in August 1998, its manufacturer submitted REMS to FDA in 2009 and obtained approval, and proposed post-marketing studies and clinical trials for five times. It can be seen that FDA has issued specific guidelines for post-marketing risk management of biopharmaceuticals to encourage multi-role participation in risk management, realize effective communication with patients, and continuously supervise the risk of biopharmaceuticals, so as to reduce the risk of the use of biopharmaceuticals. For biopharmaceuticals, China has not yet formulated systematic and specific implementation rules and guidelines, and there is still lack in post-marketing risk management. It is suggested that China can learn from the measures and system of post-marketing risk management of biopharmaceuticals in the United States, involve stakeholders in post-marketing management, enhance patients awareness of drug use risks through effective communication, and further improve the post-marketing research management system to guarantee patients safety of drug use.
KEYWORDS United States; Biopharmaceuticals; Post-marketing risk management; Enlightenment
自1982年全球首个生物技术药物重组胰岛素上市以来,经过近40年的发展,目前已有细胞因子、重组酶和激素、单克隆抗体、融合蛋白、基因治疗药、细胞治疗产品和基因工程疫苗等200多种生物药上市[1],这些生物药在癌症和遗传性疾病等诸多严重疾病的治疗中发挥着重要且关键的作用。与一般小分子化学药相比,生物药分子量大且结构复杂,其作用方式的特殊性与结构的不稳定性使得其存在潜在安全问题,尤其是免疫原性的风险较大,因而生物药上市后需要更为严格的监管,而上市后监管则是其监管的重要方式。本文以英夫利昔单抗为例,通过研究其在美国上市后的风险管理,探究美国生物药上市后的风险管理策略,为我国生物药上市后风险管理提供参考。
1 美国生物药上市后风险管理概述
药物上市后风险管理是指运用多学科方法对已上市的药品实行记载、监控、评价和干预其不良反应的过程[2]。由于通过临床试验药品的严重药物不良事件相对罕见,在临床试验中通常难以被揭示,故必须加强药品上市后管理,以及时发现临床试验中未确定的低发生率不良反应、药物长期效应以及药物相互作用等,降低其对人类健康的损害。目前,美国针对药物上市后的风险采取了多种手段进行管理,对于生物药而言,与其他药品相似,美国FDA进行上市后风险管理的措施主要包括“风险评估与降低策略(Risk evaluation and mitigation strategy,REMS)”和“上市后研究和临床试验”(Post- marketing clinical trial and study)两方面。
1.1 REMS
1.1.1 REMS的定义 REMS是美国FDA用于管理与药品相关的已知或潜在的严重风险而要求的一项程序。2007年,美国《食品和药品管理修正法案》(The Food and Drug Administration Amendments Act,FDAAA)授权FDA实施REMS,将其作为一项必要的风险管理计划,以确保药品收益大于其风险[3]。REMS旨在帮助减少药品特定严重不良事件的发生或减轻其发生的严重程度。制药企业在向FDA提交REMS后,需根据REMS中的要素(详见“1.1.2”项下)开发并提供相应培训,进行风险管理沟通以支持REMS系统顺利运作,确保医疗保健服务者(Health care providers,HCPs)、患者等遵守REMS,并定期向FDA提交REMS評估报告。而FDA在审查REMS评估报告或其他信息后,若确定不再需要REMS中的措施来确保药物的收益大于其风险,则可能会撤回已批准的REMS或删除其中的某些要素。
1.1.2 REMS的要素 REMS包括用药指南、沟通计划、确保安全使用要素、实施系统和评估报告提交时间表等5个要素内容[4]。根据FDAAA要求,REMS可包括前4个要素中的1个或多个,而评估报告提交时间表则是必备要素。
其中,用药指南是指在开具处方时向患者发放的确保药物安全有效的指导文件,其内容须经美国FDA批准,包括药品名称、重要安全信息、药品使用说明、用药禁忌证、用法用量、注意事项以及患者应当采取的预防不良事件的重要措施等。沟通计划是指申请人制定并实施的、提供给HCPs、用于告知REMS所涉及的用药安全性风险信息的工具,包括风险信息、与REMS计划操作和要求相关的信息等。确保安全使用要素是指确保药物安全和正确使用的基本要求,包括处方医师认证要求,用药患者登记要求等。为确保这些要素的实施,可以在REMS中实施系统部分提出合理的步骤与措施[5]。
评估报告提交时间表是申请人根据其制定的REMS评估计划,定期向FDA提交评估报告的时间安排。该评估报告包括申请人拟评估REMS绩效的指标、数据源和方法,以及REMS对医疗系统和患者的影响等[6]。通过审查评估报告,美国FDA将确定REMS是否实现了预期目标、其风险管理策略是否需要修改等内容。
1.1.3 实施REMS的考量要素 无论是生物药还是化学药,并不是所有药品都可以实施REMS。对于哪些药品应实施REMS,美国FDA通常会综合考虑药品创新程度、疗效和安全性等因素。具体而言,包括可能与药物有关的任何已知或潜在不良事件的严重性以及此类事件在适应证人群中的发生率、药品用于适应证的预期效果、适应证的严重程度、是否为新的分子实体、预期用药周期或实际治疗时间、估计可能使用该药品的人群数量[7]。截至2020年6月底,FDA现存批准的58项REMS中,共涉及13个生物药[8]。
1.2 上市后研究和临床试验
1.2.1 定义与类型 2007年,FDAAA第901条授权FDA对批准的药品和生物制品提出上市后研究和临床试验的要求,而对于这类“研究和临床试验”,美国《食品药品管理补充法案》(2007年)将其区分为“研究(Study)”和“临床试验(Clinical trials)”两类,并最终统一界定为药品的“上市后研究和临床试验制度”,简称为“上市后研究”[9]。具体而言,上市后研究包括“上市后要求(Postmarketing requirements,PMR)”和“上市后承诺(Postmarketing commitments,PMC)”两种:前者是指申请人需要进行的药品上市后所有必需的研究或临床试验;后者是指申请人与FDA商定后同意进行的药品上市后的研究或临床试验,其通常没有法规强制要求。此外,FDAAA授权FDA监督药品和生物制品申请人的PMR/PMC进展情况[10]。
1.2.2 PMR与PMC考量要素 在批准新药上市后,美国FDA会对与用药相关的已知的严重风险进行评估。如果已知数据提示其存在严重风险的潜在可能性,FDA将会对所批准上市药品的持有人提出进一步开展上市后研究的要求,即PMR;药品持有人需承诺开展研究,以收集有关药品安全性、有效性或最佳用药方式等信息。
需要注意的是,只有获批药品在被评估或识别为具有 “严重风险”时,FDA才会将相应要求视为PMR;未被评估或识别为具有“严重风险”的药品,在FDA与药品持有人商定同意后会将相应要求视为PMC[10]。此外,在上市后研究或临床试验过程中,FDA会要求收集特定的不良事件案例、定期汇总和评估不良事件信息、按年度提交研究进展报告等,从而加强上市后药物警戒管理,监督PMC实施。
2 以英夫利昔单抗为例的上市后风险管理要求
2.1 英夫利昔单抗简介
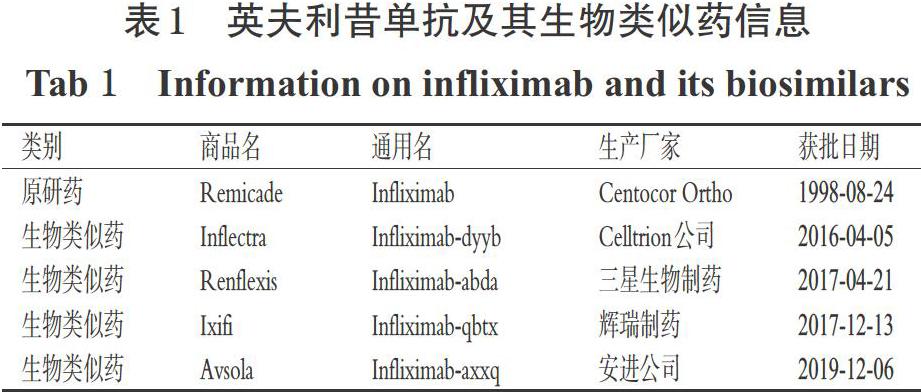
英夫利昔单抗最早由Centocor Ortho生物技术公司(该公司于1999年被强生公司收购)研发,于1998年8月经FDA批准在美国上市,商品名为类克(Remicade)。该药是一种肿瘤坏死因子(TNF)阻滞剂,通过阻断TNF的活性来抑制免疫系统,用于治疗克罗恩病、儿科克罗恩病、溃疡性结肠炎、儿科溃疡性结肠炎、类风湿关节炎、银屑病关节炎、强直性脊柱炎以及斑块状银屑病等[11]。2016年以来,FDA先后批准Celltrion公司、三星生物制药、辉瑞制药、安进公司的英夫利昔单抗类似药上市,详见表1。
英夫利昔单抗最常见的不良反应为感染(如上呼吸道、鼻窦炎和咽炎等)、输液相关反应、头痛和腹痛,发生率>10%[11-15];重大不良反应风险为严重感染(如活动性肺结核,包括潜伏性肺结核的再激活)、侵入性真菌感染(包括组织胞浆菌病、球菌病)以及细菌、病毒和其他由于机会性病原体引起的感染等。此外,该药的另一类严重不良反应为淋巴瘤及其他恶性肿瘤,已发现有報道病例[11-15]。
2.2 英夫利昔单抗的REMS
在2007年开始实施REMS后,Centocor Ortho公司于2009年11月向美国FDA提交类克的REMS并获批准,其目的在于通过提醒和警告HCPs与使用类克相关且未被识别的组织胞浆菌病和其他侵袭性真菌感染,并就此风险进行患者教育[16]。该REMS共包括用药指南、沟通计划和评估报告提交时间表3个要素。值得一提的是,对生物类似药而言,FDA要求当原研药具有REMS时,类似药也必须具有REMS,并建议与原研药共享使用同一个的REMS,否则应在类似药与原研药各自不同的REMS中具有相同的目标和要求。
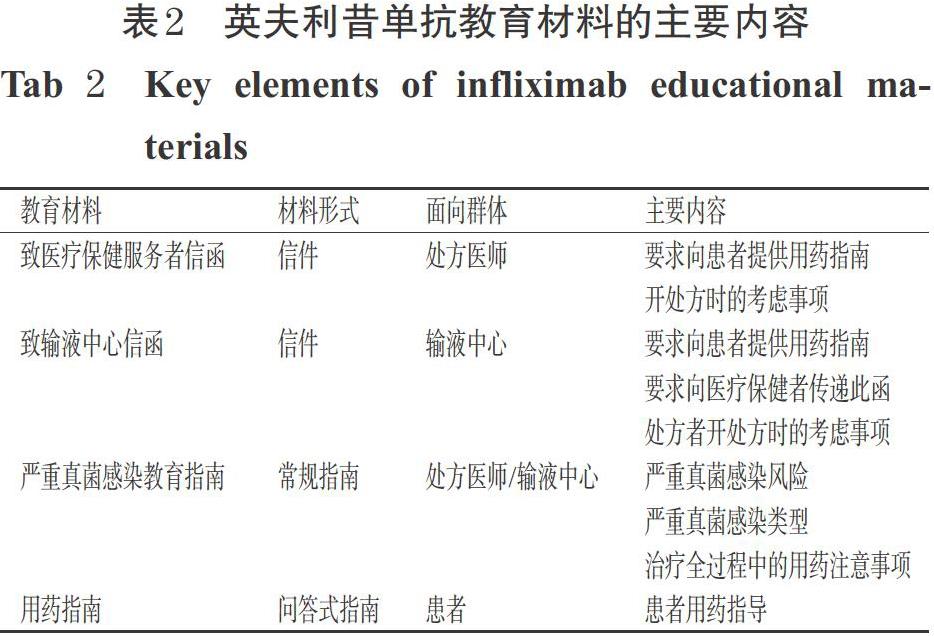
2.2.1 用药指南 类克的用药指南中列出9个主要问题,包括关于类克的最重要信息(Most important information)、类克产品介绍、不宜接受类克治疗的人群、在使用类克过程中与医师保持沟通的建议、如何使用类克、使用类克时可能出现的不良反应、使用类克时的注意事项、类克的主要成分和其他信息等。以此帮助患者了解药品以及使用注意事项、存在的不良反应和风险。例如在类克用药指南的最重要信息一栏中,强调关于该药品最重要的风险信息是导致感染和肿瘤的严重副作用;在使用前,患者需要向医师说明身体健康情况(包括患过任何类型的肿瘤),以便医师评估患者的健康状况;在使用过程中,患者需要注意时刻与医师保持必要的沟通,当患者出现任何感染的迹象时,必须马上告知医师。
REMS要求每次使用英夫利昔单抗时,医务人员都要向患者发放一份用药指南。Centocor Ortho公司需向医疗机构的输液中心(以下简称“输液中心”)提供最新的指南,并在销售包装盒上注明“向患者提供用药指南”字样。
2.2.2 沟通计划 沟通计划的对象是HCPs,主要通过REMS网站、发放教育材料等工具开展。英夫利昔单抗的网站地址为www.remicade.com,该网站主要向患者介绍类克,包括“了解类克” “继续使用类克”以及“类克的输液过程”等栏目。
根据REMS要求,Centocor Ortho公司发放的教育材料有面向医疗保健服务者、输液中心、患者等不同群体的信件和指南,主要内容均围绕使用英夫利昔单抗存在的风险和注意事项,但在面向群体、信件框架和主要内容方面存在差异,详见表2。
2.2.3 评估报告提交时间表 根据REMS计划,Centocor Ortho 公司需从REMS批准之日起18个月、3年和7年时向美国FDA提交REMS评估。并且为便于提供尽可能多的信息,同时留出合理的时间准备提交,美国FDA要求每项评估的提交时间不早于评估的提交截止日期前60天,且不晚于截止日期。
而英夫利昔单抗REMS由于其用药指南于2011年2月修订更新,后经FDA审查确定英夫利昔单抗不再需要REMS来确保其收益超过风险,故于2011年8月被FDA撤回。
2.3 英夫利昔单抗的上市后研究
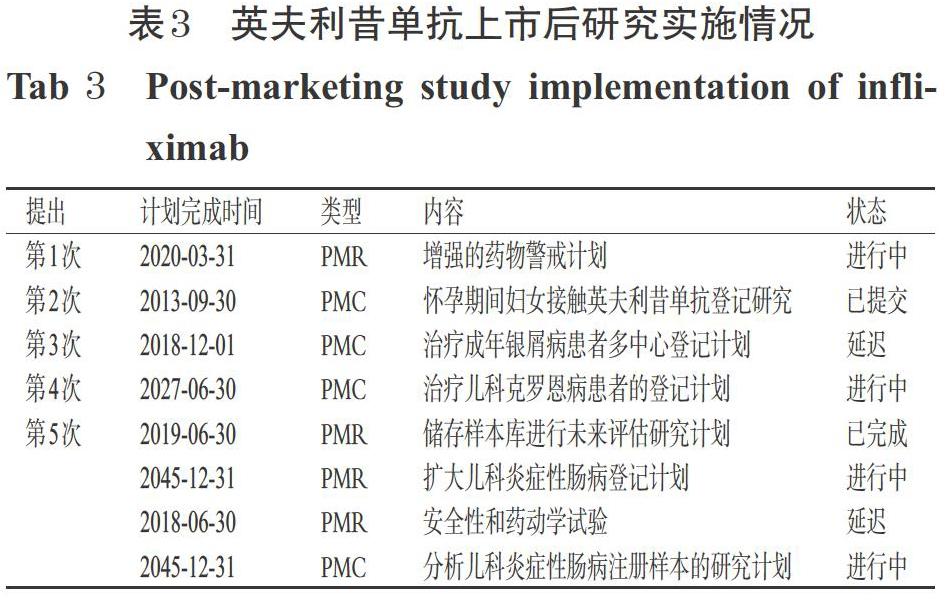
自英夫利昔单抗首次上市以来,美国FDA先后5次向Centocor Ortho公司提出PMR和PMC要求,共涉及8个方面的研究[17],详见表3。
2.3.1 第1次PMR FDA第1次提出的PMR是增强药物警戒管理,用于报告使用英夫利昔单抗治疗的儿童、青少年和青年人(年龄不超过30岁)发生恶性肿瘤不良反应的信息。FDA要求Centocor Ortho公司主动询问不良反应报告者以获得与恶性肿瘤诊断有关的未知临床信息,同时要求加快向FDA报告初始和后续信息。该项研究计划长达10年,以收集、分析数据从而确定恶性肿瘤发生的风险。该项研究原本预计2020年3月完成,但目前仍正在进行中。
2.3.2 第2次PMC FDA第2次提出的研究为PMC,主要针对患有克罗恩病、类风湿性关节炎和银屑病关节炎的孕妇,对其在怀孕期间使用英利昔单抗的情况进行前瞻性和观察性的登记研究,评估了使用英夫利昔单抗的妊娠风险。该项研究结果已于2013年提交。
2.3.3 第3次PMC 这一次,Centocor Orth公司同意在美国进行一项前瞻性、多中心的注册,主要针对4 000名在美国使用英夫利昔单抗的成人银屑病患者。研究计划收集有关患者特征、人口统计学等数据,以描述和评估包括严重感染、结核病、自身免疫反应和死亡等严重不良事件以及其他不良事件的发生率。目前,该项研究由于患者招募困难被推迟,但仍在进行中。
2.3.4 第4次PMC 在FDA第4次提出的PMC中,Centocor Ortho公司针对0~19岁的克罗恩病儿童患者,通过设计和实施正在接受英夫利昔单抗治疗的患者登记表,收集人口统计学、疾病特征、同期用药史等信息,以获得长期临床疗效和安全性信息。目前,该项研究计划正在进行中,预计2027年完成。
2.3.5 第5次PMR和PMC FDA第5次共提出的4项研究计划,具体如下:第1项PMR通过调查患者病理样本进行长期评估的研究,以确定易使炎症性肠病患者发展为肝脾T细胞淋巴瘤的基因突变和其他生物标志物,该研究于2019年完成但尚未提交结果;第2项PMR是扩大儿科炎症性肠病登记,计划将溃疡性结肠炎和不明原因结肠炎的儿科患者包括在内,该计划目前正在进行中,预计2045年12月完成;第3项PMR是安全性和药动学试验,以评估儿科溃疡性结肠炎和儿科克罗恩病患者最低用药剂量,目前该研究由于入组人数过少而处于延迟状态;第4项是PMC,计划使用新测定方法分析来自儿科炎症性肠病登记样本,目前该研究正在进行中,并预计2045年12月完成。
3 美国生物药上市后风险管理特点
与小分子化学药不同,生物药的不良反应更多由其药效学效应和个体免疫反应引起,故生物药的潜在不良反应更频繁地涉及血栓、感染、全身炎症反应和良性、恶性或不确定性肿瘤等[18]。故相对于化学药,美国在生物药上市后风险的监测中也更为严格,持续时间也更长。
3.1 多角色参与风险管理
从FDA实行的REMS来看,美国生物药上市后风险管理由多角色参与,包括政府部门、制药企业、输液中心、医疗保健服务者以及患者等多个利益相关方。其中,FDA制定相应规定和指南,由药品评估与研究中心(Center for Drug Evaluation and Research,CDER)的风险管理部(Division of Risk Management,DRM)协调生物制品评估和研究中心(Center for Biologics Evaluation and Research,CBER)统筹生物药上市后风险管理,包括所有REMS的评审、修订和评估等[19];制药企业承担其产品上市后风险的教育和培训责任,提供教育材料,以提高用药者对生物药使用风险的认识水平;输液中心每当使用药品时都需要向患者发放用药指南,确保患者对所用药物的充分了解以及对生物药风险性的警觉;HCPs保持与患者的充分沟通,提供生物药的咨询途径,以便及时发现不良反应,降低药物风险的损害。而与化学药不同的是,生物药的REMS中致力于登记所有患者,要求尽可能确保每一位患者都注册参加REMS计划,以进一步支持生物药长期的安全使用研究。
3.2 与患者有效沟通
美国FDA通过用药指南向患者传达生物药的信息和相关注意事项,以确保患者对所用药物有一定认识;同时,要求制药企业通过REMS,确保HCPs和患者在整个用药过程时刻保持沟通,HCPs须向患者说明生物药的特殊性,以及生物药的潜在风险等相关信息。值得一提的是,与化学药不同,由于生物药在免疫原性方面存在的风险,故在与患者沟通的过程中,REMS更加强调对患者的定期检查,以便时刻关注患者的自身免疫、感染以及过敏等情况。此外,HCPs需提供有效的患者咨询途径,并提醒患者用药过程中的注意事项,了解患者用药后情况,及时监测并报告患者出现的不良反应。同时,制药企业需建立专门的产品网站,提供联系方式和患者反馈系统,通过多途径的沟通交流,以降低不良反应的发生风险,支持REMS系统的运作。
3.3 加强上市后研究,进行持续监测和评估
笔者通过对2016-2020年美国FDA批准的化学药和生物药开展上市后研究的情况进行统计比较后发现,相较于化学药平均5 423天的上市后研究持续周期,生物药上市后研究平均周期为6 507天,可见对生物药的上市后风险更强调持续、长期的监测和管理。而FDA通过要求生物药上市申请人实施上市后研究,可以有效弥补新药上市时存在未被发现的风险的缺陷;另一方面,通过评估已知的严重不良反应风险,对相应不良事件加强监测,可以及时发现或预防严重不良反应的发生,确保患者用药安全。并且生物药上市申请人每年按时提交研究进展报告,可进行长期持续监测与评估,以降低生物药的潜在风险。此外,FDA在REMS中亦要求申请人定时提交风险评估报告,以保证对上市生物药长期动态的监管。
4 我国生物药上市后风险管理的现状与改进
4.1 我国上市后风险管理现状及问题
2019年,我国新修订的《药品管理法》中第七十七条提出,“药品上市许可持有人应当制定上市后风险管理计划,开展上市后研究,加强持续管理”[20]。在其配套规章《药品注册管理办法》第三十九条中规定,“药品审评中心必要时需在药品注册证书的附件中附药品上市后研究要求”[21]。
在宏观层面,尽管我国通过法律法规对药品上市后管理做出了要求,但目前我国上市后风险管理活动主要集中在药物警戒制度的建设,对于上市后研究的具体要求,我国尚未制定系统、具体的实施细则和指南。在微观层面,由于我国医患之间因医药专业性导致的沟通困难以及信息不对称问题仍然十分严重,制药企业与医疗服务提供方进行有关产品的学术交流较少等问题的存在,我国目前的医疗卫生体系仍缺乏覆盖制药企业、医院、药师、患者等在内的沟通机制与平台,难以从微观层面做好上市后风险管理。尤其对于生物药而言,其在我国上市时间较短,且正处于密集研发与申报的阶段,对于其上市后管理的要求,应积极发挥各利益相关者作用,建立上市后安全风险管理与评价的科学体系。
4.2 美国相关管理经验的启示及相应建议
4.2.1 鼓励多方参与风险管理,及时报告不良反应 对生物药的风险进行上市后评估需要各种各样的数据来源,包括从患者登记到就诊的记录以及纸质或电子治疗数据等。目前,我国正在建立以药品上市许可持有人(MAH)为主体的上市后管理和药物警戒管理体系。在当前生物药产业的快速发展阶段,单抗类药物、生物类似药等陆续上市,而在医疗机构、患者等对生物药的认知度不高的背景下,建议我国规定MAH在承担主体责任的同时,应尽可能吸纳利益相关方参与上市后管理。一方面,持有人可就上市生物药用药特殊性与安全性风险,针对医疗机构、医务人员、零售药店、患者等开展用药教育和培训,提高各方对生物药用药特性的理解及对其潜在风险的认识。另一方面,我国药品监督管理部门可借助医疗机构资源,开展生物药用药处方以及用药患者等信息登記管理,加强患者用药回访管理,保障用药记录的可追溯性,以便及时发现并报告用药风险和不良反应。
4.2.2 建立有效沟通途径,提升患者用药风险意识 患者作为药品使用者,受药品安全风险的直接影响。因此,美国FDA一直强调保障患者在上市后风险管理中的参与度与权益。为更好地在我国实施生物药上市后风险管理,建议MAH可以建立并维护生物药专题网站,公开药品详细信息,包括说明书、用药指南、注意事项,甚至严重不良反应警告等,以供公众自主查阅学习。此外,建议MAH在网站上提供直接沟通途径,以获得及时的反馈。
另一方面,我国需要促进医患双方对生物药可能存在的风险开展有效沟通,并提供患者用药咨询的途径,保障患者对所用药物的知情权和所用药物存在风险的知晓,提高患者的用药依从性。同时,我国还需针对生物药上市后风险进行专门化管理,通过发放生物药专药专用的用药指南或宣传手册等方式,鼓励患者了解所使用药物,提高患者风险意识并进行自我用药管理。
4.2.3 推进上市后研究,加强持续监测与管理 目前我国药物警戒制度仍在建设中,但药物警戒活动主要围绕不良反应监测开展工作,在上市后风险管理方面仍有所欠缺[22],且尚未建立专门针对生物药上市后风险管理的法规或指南。建议我国进一步完善上市后研究管理制度,对新上市生物药的临床用药安全性进行及时监测,与MAH保持沟通并提出研究要求。在此过程中,需要制定详细的配套技术指南以提供行业指导,并建立信息公开制度,将相关研究信息通过网站等途径进行公开,提高公众对生物药临床使用和上市后研究的认知度,促进相应研究的顺利进行。
5 结语
美国生物药上市后的风险管理措施主要采取REMS和“上市后研究和临床试验制度”两种方式,其中后者包括PMR和PMC两类。美国FDA通过出台具体指南,鼓励多角色参与,以实现与患者的有效沟通,对生物药风险进行持续监管。而我国在生物药上市后的风险管理方面仍存在欠缺,故可借鉴美国相关经验和措施。建议我国尽可能吸纳利益相关方参与上市后风险管理,建立与患者开展有效沟通的机制与平台,提升患者用药风险意识,并且进一步完善上市后研究管理制度,保证对生物药用药风险的持续监管,保障患者用药安全。
参考文献
[ 1 ] 王军志.我国生物药监管科学的发展概述[J].中國新药杂志,2018,27(21):2465-2471.
[ 2 ] 竟永华,郭剑非,李行.美国药品风险管理指南与案例分析[J].中国药物警戒,2005,2(4):193-196、200.
[ 3 ] FDA. Best practices in drug and biological product postmarket safety surveillance for FDA staff[EB/OL]. (2019- 11-06)[2020-06-26]. https://www.fda.gov/media/130216/download.
[ 4 ] FDA. A framework for benefit-risk counseling to patients about drugs with a REMS[EB/OL]. [2020-06-26]. https://www.fda.gov/media/107591/download.
[ 5 ] SCHNEIDER P J. Risk evaluation and mitigation strategies[J]. J Nurse Pract,2012,8(9):747-748.
[ 6 ] FDA. REMS assessment:planning and reporting[EB/OL]. (2019-02-04)[2020-07-03]. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/rems-assessment-planning-and-reporting.
[ 7 ] FDA. REMS:FDAs application of statutory factors in determining when a REMS is necessary[EB/OL]. (2019- 04-04)[2020-06-26].https://www.fda.gov/regulatory-information/search-fda-guidance-documents/rems-fdas-application-statutory-factors-determining-when-rems-necessary-guidance-industry.
[ 8 ] FDA. Approved risk evaluation and mitigation strategies (REMS)[EB/OL].[2020-06-26].https://www.accessdata.fda.gov/scripts/cder/rems/index.cfm.
[ 9 ] 罗雪燕,赖寒,陈绍成,等.美国药品上市后研究的监管制度及其对我国的启示[J].中国药房,2017,28(31):4330- 4334.
[10] FDA. Postmarketing studies and clinical trials :implementation of section 505(O)(3) of the FDCA[EB/OL]. (2011-04-11) [2020-06-26].https://www.fda.gov/regulatory-information/search-fda-guidance-documents/postmarketing-studies-and-clinical-trials-implementation-section- 505o3-federal-food-drug.
[11]FDA. Remicade medication guides[EB/OL]. (2020-05-14) [2020-06-26]. https://www.accessdata.fda.gov/drugsatfda_ docs/label/2018/103772s5385lbl.pdf#page=48.
[12]FDA. Renflexis medication guides[EB/OL].(2019-06-26)[2020-06-26]. https://www.accessdata.fda.gov/drugsatfda_ docs/label/2019/761054s009lbl.pdf#page=52.
[13]FDA. Inflectra medication guides[EB/OL]. (2019-06-18)[2020-06-26]. https://www.accessdata.fda.gov/drugsatfda_ docs/label/2019/125544s009lbl.pdf#page=59.
[14] FDA. Ixifi medication guides[EB/OL]. (2020-01-16)[2020-06-26]. https://www.accessdata.fda.gov/drugsatfda_ docs/label/2020/761072s006lbl.pdf#page=43.
[15] FDA. Avsola medication guides[EB/OL]. (2019-12-06)[2020-06-26]. https://www.accessdata.fda.gov/drugsatfda_ docs/label/2019/761086s000lbl.pdf#page=48.
[16] FDA. Remicade REMS document[EB/OL]. (2011-02-17)[2020-06-26]. http://www.accessdata.fda.gov/drugsatfda_ docs/rems/Remicade_2011-02-17_REMS_Document.pdf.
[17] FDA. Postmarket requirements and commitments[EB/OL]. [2020-06-26].https://www.accessdata.fda.gov/scripts/cder/pmc/index.cfm?StartRow=1&StepSize=1&Paging=Yes#.
[18] INGRASCIOTTA Y,CUTRONEO P M,MARCIAN? I, et al. Safety of biologics,including biosimilars:perspectives on current status and future direction[J]. Drug Saf,2018,41(11):1013-1022.
[19] 鮑程程,王宏伟,杨悦. FDA风险评估与减低策略实施与借鉴的思考[J].中国药物警戒,2017,14(5):283-288.
[20] 全国人民代表大会.中华人民共和国药品管理法[S/OL]. (2019-08-26)[2020-06-26]. https://www.nmpa.gov.cn/xxgk/fgwj/flxzhfg/20190827083801685.html.
[21] 国家市场监督管理总局.药品注册管理办法[S/OL]. (2020- 01-22)[2020-06-26]. https://www.nmpa.gov.cn/xxgk/fgwj/bmgzh/20200330180501220.html.
[22] 胡歆雅,梁玉清,曾亚莉,等.中美药物警戒制度的比较研究[J].中国合理用药探索,2020,17(2):21-25.
(收稿日期:2020-09-25 修回日期:2021-01-21)
(编辑:刘明伟)