Na||Sb-Pb-Sn 液态金属电池电极的价电子结构与热-电性能计算*
2021-05-06张健王心桥苏彤陈英郭永权
张健 王心桥 苏彤 陈英 郭永权
(华北电力大学能源动力与机械工程学院, 北京 102206)
1 引 言
可再生能源技术的快速发展, 要求研究低成本、长寿命和大容量储能系统, 以增强电网的稳定性、可靠性和安全性[1].传统的储能系统主要采用铅酸电池蓄电, 但电池循环寿命短, 充放电速度慢,且对环境污染大; 锂离子电池具有高储存能量密度, 但制造成本较高, 充、放电性能差, 电池均一性和安全性差; 液流电池结构比较复杂, 需要对电解液、离子交换膜等关键材料进行深入的技术开发;钠硫电池所采用的固体电解质的热稳定性较差、安全性差[2-5].这些电化学储能电池的主要问题是使用寿命短、成本高, 无法满足市场需求.针对这一问题, 美国麻省理工学院的Sadoway 教授提出“液态金属电池(liquid metal battery)”的新概念[6,7].实验研究表明: 液态金属电池具有成本低廉、寿命长、放电速率高、储能密度大等优点, 可以使用15 年以上, 满足大规模静态储能的要求[8-12].液态金属电池的电极材料和电解质均为液态, 工作温度范围为500—700 ℃, 阴极选用活性较高的液态碱金属或碱土金属[13].由于液态金属电池电极的工作温度比较高, 适用性受限.通过元素的添加, 使金属电极合金化, 可以降低电极的熔点, 进而降低运行成本和工作温度[14].钠与锂同族, 具有与锂相似的物理和化学性能, 钠在地壳中含量丰富、成本低.因此, 选取金属Na 作为阴极材料, 并设计在Na电极中添加熔点低的IA 碱金属元素K, Rb, Cs 等元素来降低工作温度[15,16].阳极选择Sb, 设计添加低熔点的IV 族金属Pb 和Sn, 实现阳极熔点的降低.本文设计的液态金属电池的体系是Na||Sb-Pb-Sn.应用理论研究电池合金化后的电子结构和物理性能.
随着计算材料科学的发展, 材料的理论研究有助于材料成分设计及其性能的提高[17-19].固体与分子经验电子理论在材料的电子结构和性能研究方面, 具有丰富的研究成果, 涉及材料的力学性能、热性能、电磁性能和光特性等方面.性能的理论计算与实验相符[20-22].其优势在于: 理论模型简单, 没有涉及复杂的微积分及数理方程计算, 而且计算参数少.
本文应用固体与分子经验电子理论研究液态金属电池Na||Sb-Pb-Sn, 并通过价电子结构的分析和热、电性能(电极熔点, 结合能, 电势, 开路电压)计算, 揭示电池性能与电子结构的关联性.
2 理论模型
固体与分子经验电子理论是基于能带理论和Pauling 的价键理论基础之上, 研究物质性能与其价电子结构的关联性[23-30].计算物理性能包括熔点、结合能、磁矩、居里温度、超导转变温度及光吸收带宽等[20,21,31], 性能的理论值与实验相符.理论包括三种假设及一种分析方法(键距差法).
2.1 价电子结构的三种假设
2.1.1 双态的假设
电子结构存在两种基本的状态: 能量低的稳态(h 态)及能量高的激发态(t 态), 双态分布着各种各样的电子, 如: s, p, d 轨道上的价电子ns,np,nd; 共价电子nc; 晶格电子nl; 哑对电子nY; 磁电子m3d及总电子nT.
2.1.2 杂化态叠加的假设
依据量子力学态叠加原理, 物质的电子结构是由h 态和t 态成分比例分数Ch和Ct的叠加.其比例分数Ch和Ct是不连续的, 呈现量子化.其计算公式为
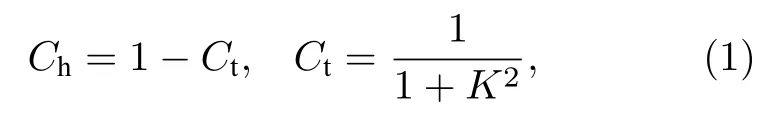
在h 态,Ct= 0,K=∞.t 态时,Ct= 1,K= 0.在给定原子杂化状态下,K的取值为

其中,l,m,n,l',m',n'分别表示h 态和t 态中的s,p, d 价电子和晶格电子数.当(2)式h 态的价电子全部是晶格电子时不适用, 即l+m+n= 0, (2)式无意义.此时的K公式修正为
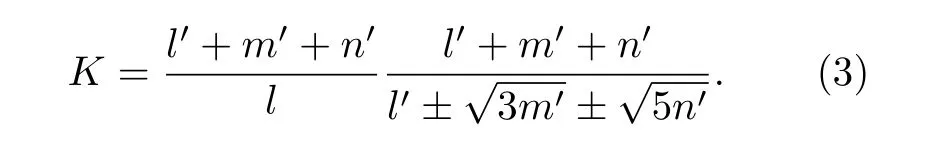
各种电子数可以通过h 态和t 态成分比例分数计算.并通过双态的加权平均方法计算出各种电子数, 这些电子数与其电子态(又称杂化态)一起组成元素的价电子结构的杂化表.本文研究的阴、阳极金属元素的杂化表, 见附录A.
2.1.3 键距方程
对于晶胞内任何两个相邻的u 和v 原子所形成的u—v 键, 其键矩可以根据Pauling 提供的键距计算公式:
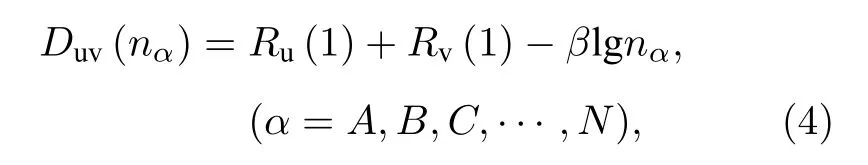
其中,Duv(nα)为键距,nα是共价电子对数,Ru(1)和Rv(1)分别代表u, v 原子的单键半矩.参数β取值如下:

其中,ε是偏离量, 取值范围0 <ε< 0.050;nM是共价电子对数nα中的最大值.
2.2 键距差方法[20,21]:
第1 条键:

第i条键:
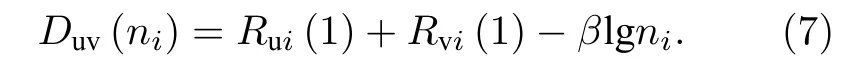
(7)式—(6)式则有:
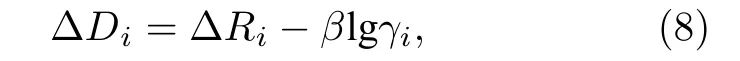
其 中: ΔDi=Duv(nA)-Duv(ni) ;ΔRi=Ru(1)+Rv(1)-Rui(1)-Rvi(1);γi=nA/ni.由(8)式得
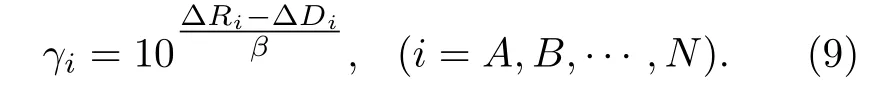
晶胞中总的共价电子数nc:
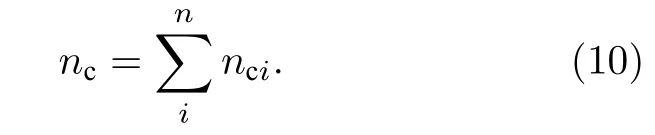
总的共价电子数也是所有等价键上的共价电子对数之和:
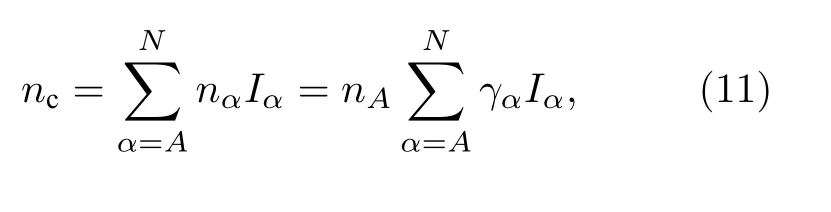
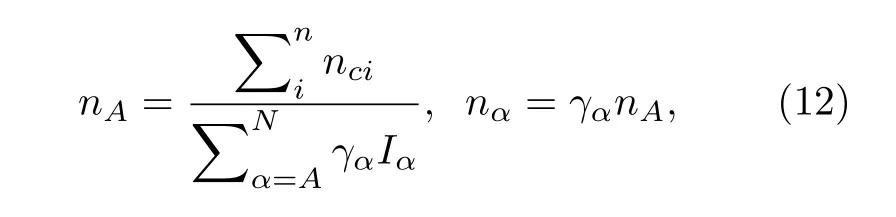
其中,Iα是等价键数, 其与结构式中的原子个数IM,原子配位数IS及成键原子的异同因子IK有关(IK的取值: 对于同类原子取1, 异类原子取2).计算公式如下:
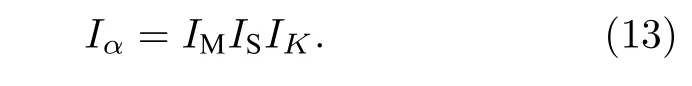
将(12)式代入(4)式, 则可以计算出u, v 原子之间的理论键距
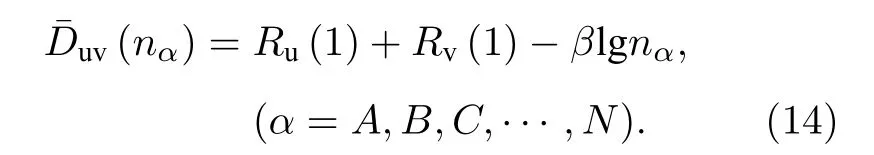
理 论 值 与 实 验 值 之 差 ΔD=(nα)-Duv(nα)作为价电子态的选择判据, 在一级近似下,|ΔD|≤0.05 Å.
2.3 性能计算模型
根据价电子结构参数, 建立结合能理论模型.能量由四项组成: 键能、晶格电子电位能、磁能及内壳层电子的内聚能.结合能的计算公式如下:

其中:b是电子对核电荷的屏蔽系数, 取值为b=313.95/(n— 0.36·δ) kJ· Å /mol,n和δ是由于原子的内层电子的屏蔽、电子的库仑、交换及关联作用对键能影响的总效应,n= 1, 2, 3, 4, 5, 7, 13,δ=2, 1, 0;f是成键因子;nl是晶格电子数;f′为晶格电子的成键因子;为等效键距;α= 0.1542;m3d是原子的磁电子数;C= 0.907P,P的取值对应于B 亚族的V, VI, VII 各族, 第VII 族的Fe, Co, Ni, 以及IB 和IIB 族元素分别依次为6, 5, 4, 3, 2, 1 和0 (0 对应IB 和IIB 族);[25].由于研究的电极金属都是主族金属元素, 没有过渡簇金属元素, 因此,结合能只有前两项.
熔化模型: 热声子能量随着温度的升高增加.当达到温度达到临界值(熔点Tm)时,n摩尔的热声子能量刚好与最强键的能量Es相等.即
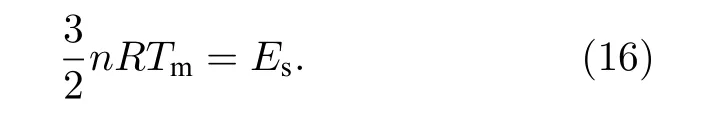
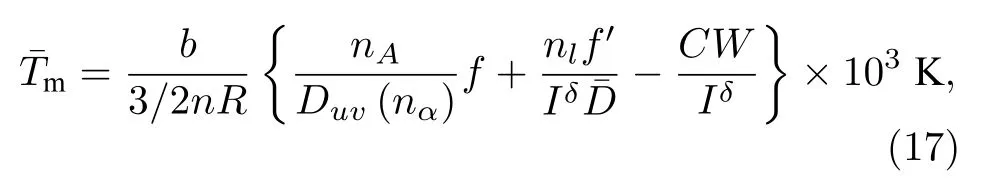
开路电压V: 对于液态金属电池而言, 开路电压等于阳、阴电极的电势差.应用阳、阴电极的电位能差除以其总的晶格电子数nl后获得, 如(18)式所示.
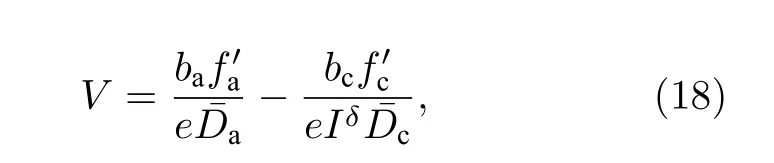
其中,e为电子电量, 下标a 与c 分别对应阳极和阴极.
3 计算分析和讨论
3.1 阴极的价电子结构与热电性能计算
为了降低电池阴极的熔点, 在阴极Na 金属中添加熔点低的IA 族碱金属元素K, Rb, Cs 形成Na1-xIAx合金电极(x≤ 0.05).Na 具有体心立方结构, 空间群为IA 族碱金属元素的微量掺杂不影响Na 的晶体结构, 等效于Na 的固溶体合金.由于原子半径的差异, 掺杂会引起Na 的点阵常数变化.应用平均原子模型计算合金的点阵常数:
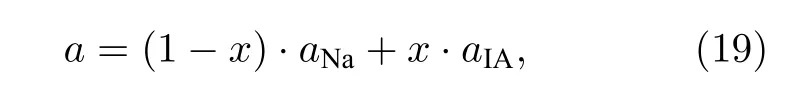
其中:aNa和aIA分别代表Na 及其他IA 族的点阵常数.合金的共价电子数nc、晶格电子数nl及平均的单键半矩R(1)分别可以通过Na 和添加IA 簇金属按成分加权平均的方法获得, 如下所示:

Na1-xIAx合金的价电子结构计算结果如表2所示.基体金属Na 取第2 杂阶, 掺杂金属K, Rb,Cs 取第4 杂阶.随着IA 族元素的掺杂量的增加,共价电子数nc, s 与p 价电子数ns与np趋于增加,晶格电子数nl减少.将这些价电子结构参数代入计算性能模型, 计算合金的热、电性能.计算结果见表3.

表1 Na1-xIAx 合金键距计算Table 1.Calculation of bond distances of Na1-xIAx alloy.
计算结果表明: 阴极合金Na1-xIAx的熔点与结合能随着IA 族原子掺杂量x的增加趋于降低;Cs 原子掺杂降低更显著.其电位能和熔点变化与结合能变化情况相似, 随着K, Rb, Cs 原子掺杂量的增加而降低.当掺杂量相同时, Cs 的Ec最低,K 时Ec最高.阴极合金的电势变化情况与熔点和结合能一致.计算结合能与实验值相符.实验值由Na 和IA 族元素的单质的内聚能[32]按成分比例平均获得:
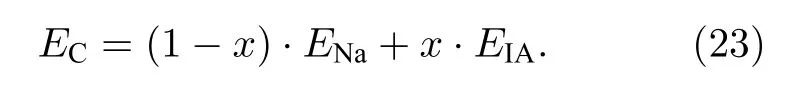
金属电极的性能与价电子结构密切相关联, 阴掺杂IA 族元素形成合金Na1-xIAx可以调控阴极的电子结构, 进而影响性能.图1 展示阴极熔点与电子结构及掺杂量之间的关联性.随着掺杂量的增加, 在Na1-xRbx和Na1-xCsx合金的熔点降低,Cs 掺杂的合金降低更明显.阴极合金的价电子结构的变化直接反应其相关性, 随着掺杂量的升高,晶格电子数nl明显降低, 共价电子nc和s 价电子数ns呈现增加的趋势.由于总电子数恒定, 晶格电子数的减少, 共价电子数的增加.等效于晶格电子向价电子的转化.根据熔化模型, 熔点的降低主要源于第二项晶格电子贡献的降低.说明热声子优先破坏非成键的晶格电子在晶格空间的分布, 降低其电位能.阴极合金的结合能也表现出相同的现象,如图2 所示, 随着掺杂量的增加, 三种合金的结合能都明显下降.Cs 掺杂合金降幅更大.从价电子结构变化看: 随着掺杂量的升高, 晶格电子数nl明显降低.说明晶格电子电位能的降低是主要原因.阴极合金电势的变化与前两者相同, 呈现下降的趋势, 如图3 所示.主要原因还是晶格电子的减少,共价电子的增加.
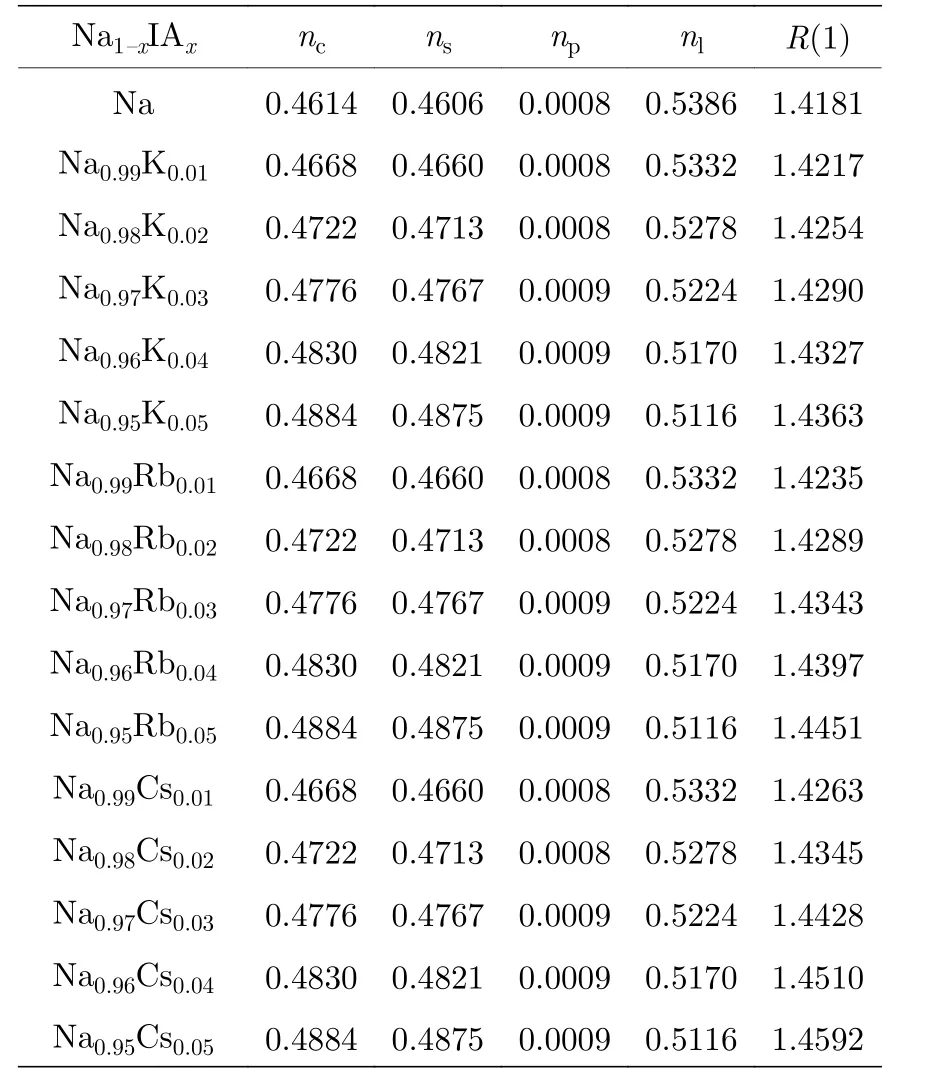
表2 Na 阴极合金的价电子结构Table 2.Valence electron structures of cathode Na based alloy.

表3 阴极Na1-xIAx 合金的熔点、结合能与电势Table 3.Melting point, cohesive energy, and electric potentials of cathode Na1-xIAx alloy.
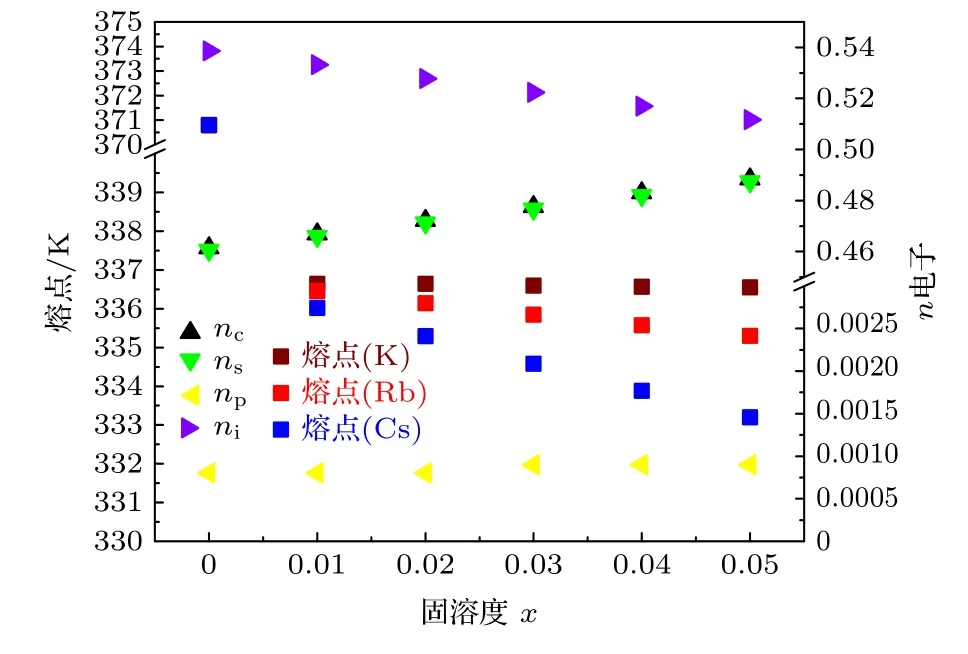
图1 阴极Na1-xIAx 合金的n 电子数与熔点的关联图Fig.1.Correlations between various electron numbers and melting points of Na1-xIAx cathode alloys.
3.2 正极合金价电子结构与热、电性能计算
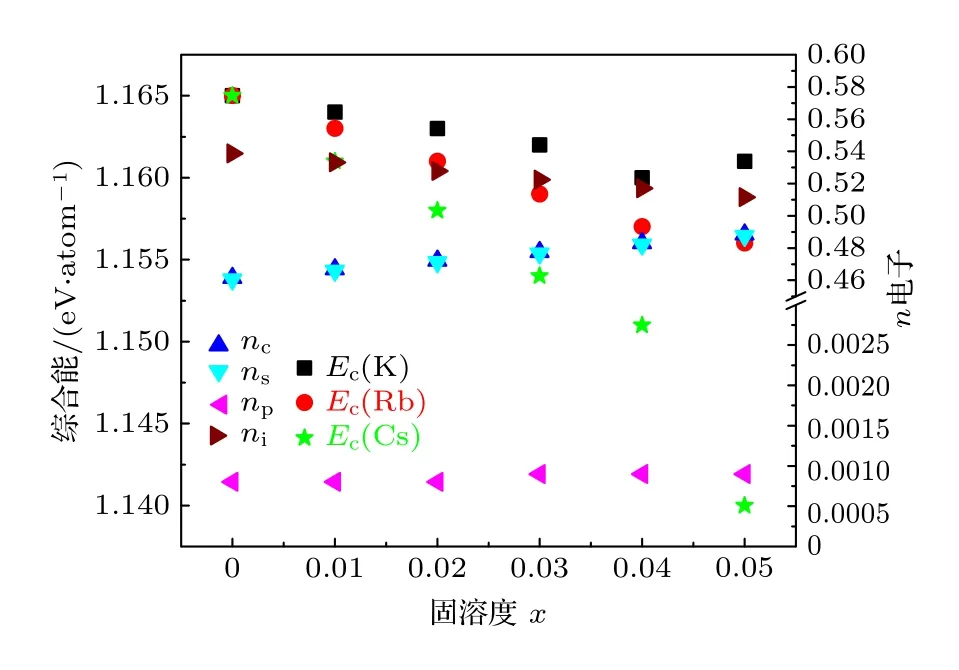
图2 阴极Na1-xIAx 合金的各种电子数与结合能的关联图Fig.2.Correlations between various electrons and cohesive energy of Na1-xIAx cathode alloys.
图3 阴极Na1-xIAx 合金的各种电子数与电势的关联图Fig.3.Correlations of various electrons and electric potentials for Na1-xIAx anode alloys.
本文选择Pb, Sb, Sn 三种元素来设计液态金属电池的阳极合金; Sn, Pb, Sb 相对于其他金属,具有较高的电负性, 适合作为液态金属电池正极材料[33-34].Na 离子输运到阳极, 与阳极金属Pb, Sb,Sn 反应形成阳极产物(合金化合物): Na3Sb, NaSn,Na15Sn4, NaPb.四种合金化合物的晶体结构, 见表4.
应用键距差方法计算这四种合金化合物中的所有键距, 计算值与实验相符, 最大键距差都满足判据: |ΔD| ≤ 0.05 Å.计算结果见表5.
四种阳极合金的价电子结构计算结果如表6所示.Na3Sb 合金化合物: 2c晶位的Sb 取第2 杂阶, 只提供价电子; 2b晶位的Na 取4 杂阶, 只提供价电子; 4f晶位的Na 取第2 杂阶, 提供晶格电子.NaSn 合 金 化 合 物: 晶 格 电 子 由16e晶 位 的Na 及32g晶位的Sn 所提供.Sn 也提供价电子, 然而位于16f晶位的Na 只提供价电子.Na15Sn4合金化合物: 晶格电子由16c晶位的Sn 及48e晶位的Na 所提供, 位于12a晶位的Na 只提供价电子.NaPb 合金化合物: 位于32g晶位的Pb 提供价电子和晶格电子; 16e晶位的Na 只提供价电子; 位于16f晶位的Na 只提晶格电子.
依据电子结构参数, 计算阳极合金化合物的热、电性能, 理论熔点与实验相符.计算结果由表7所示.计算结果表明: 四种阳极合金化合物的熔点的范围在604.89—1142.96 K 之间, NaPb 的熔点最低, Na3Sb 的熔点最高.然而电势, Na3Sb 最高1.152 V, NaSn 的最低0.7343 V.
3.3 液态金属电池的开路电压
应用开路电压(18)式系统地计算阴、阳电极之间的开路电压.表8 列出金属电极及其开路电压值.计算结果表明: Na||Sb 电极之间的开路电压最高略高于1 V.Na||Sn 电极的开路电压低, 约为前者的一半.开路电压与平均每原子的晶格电子数成反比, 平均晶格电子数最低的Na3Sb(0.2693), 其开路电压最高; 而平均晶格电子最高的NaSn, 其开路电压则最低.
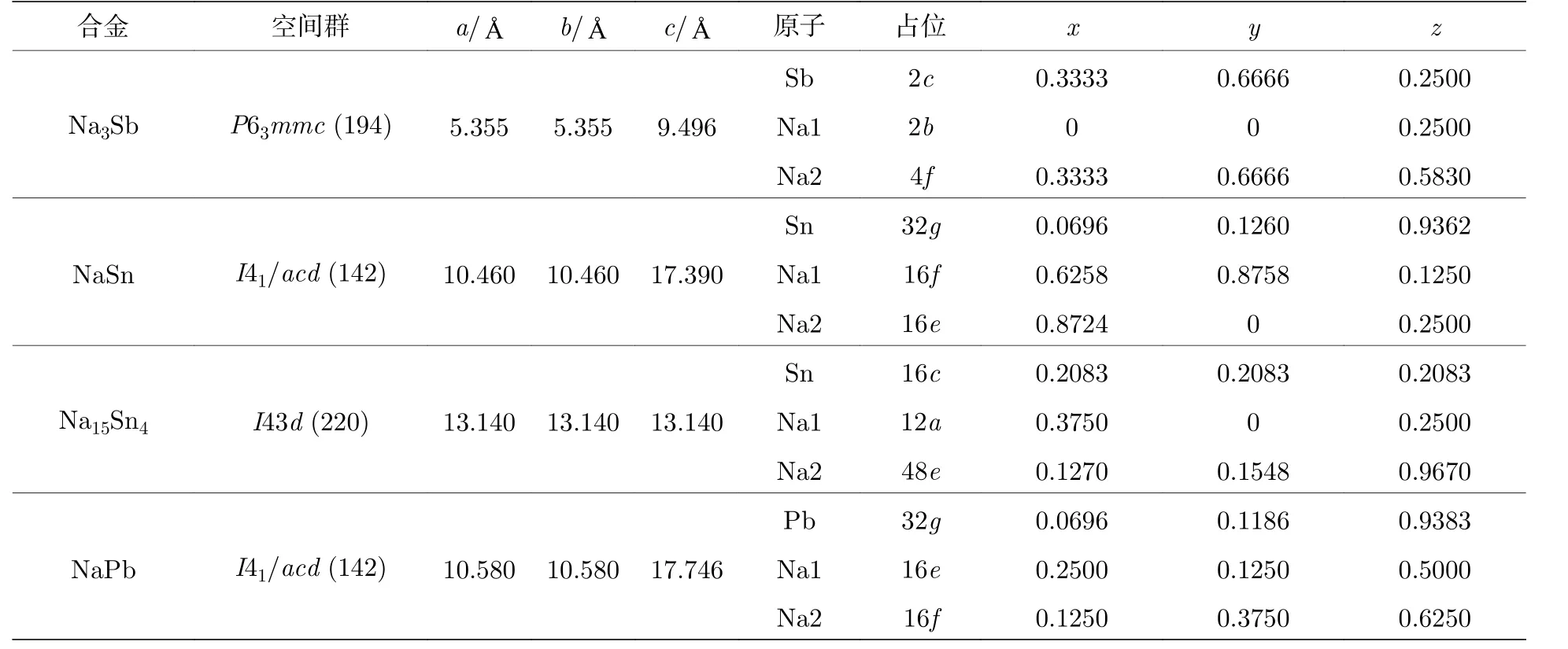
表4 正极合金的晶体结构Table 4.Crystal structures of anode alloys.
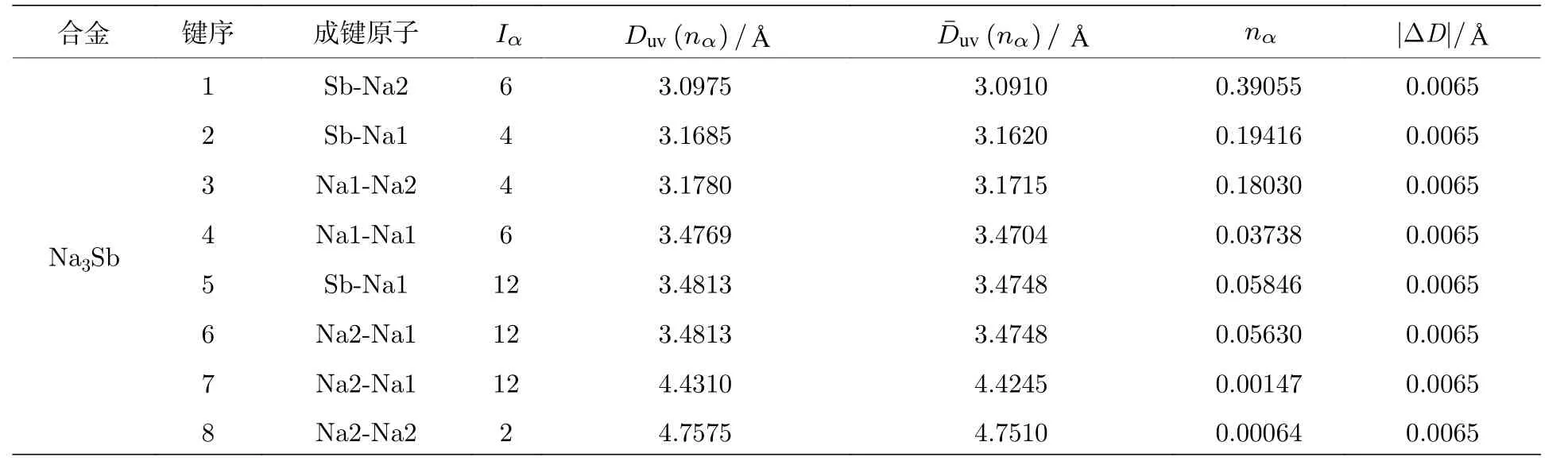
表5 阳极合金的键距Table 5.Bond distances of the anode alloy.
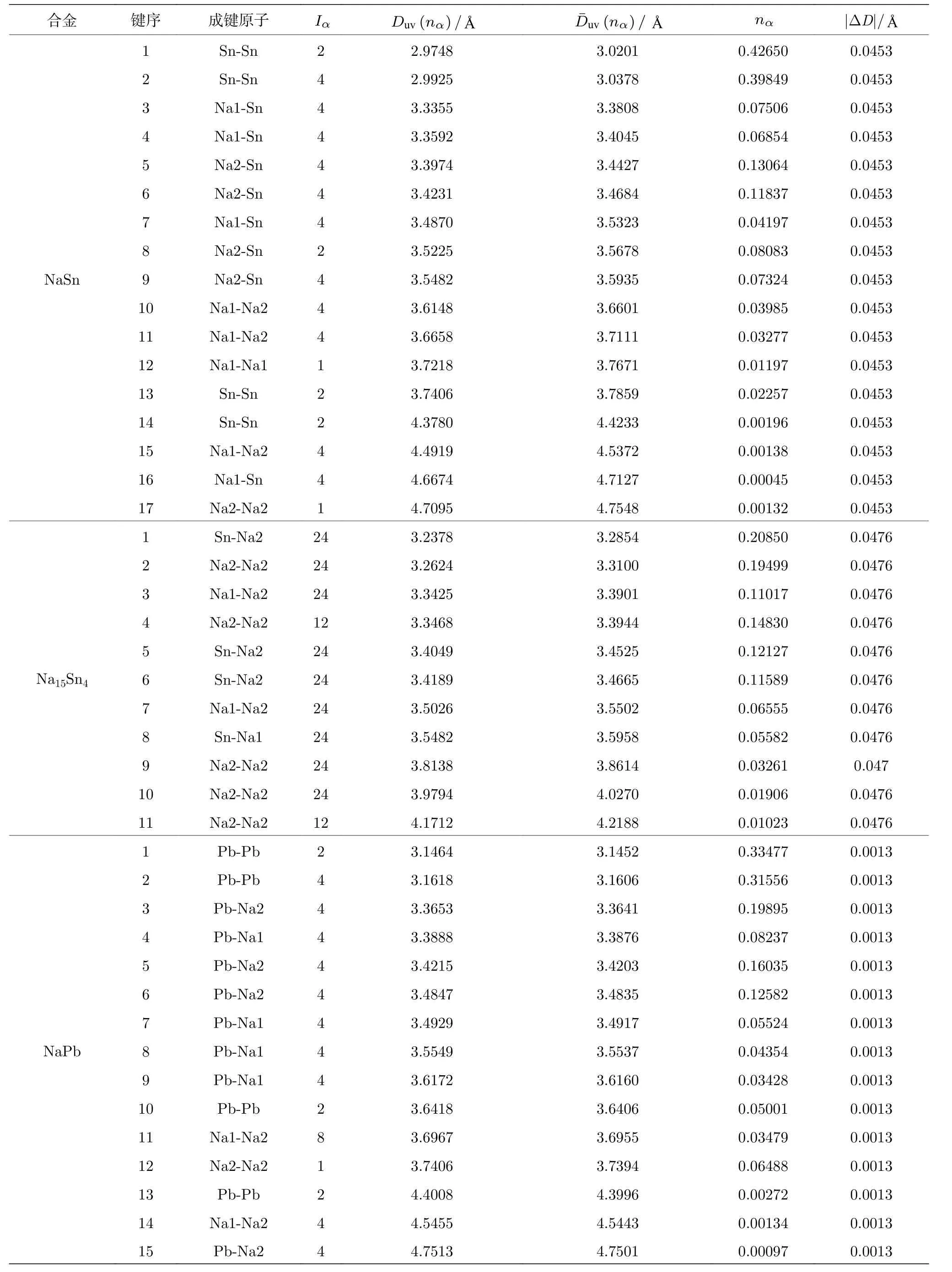
表5 (续) 阳极合金的键距Table 5 (continued).Bond distances of the anode alloy.
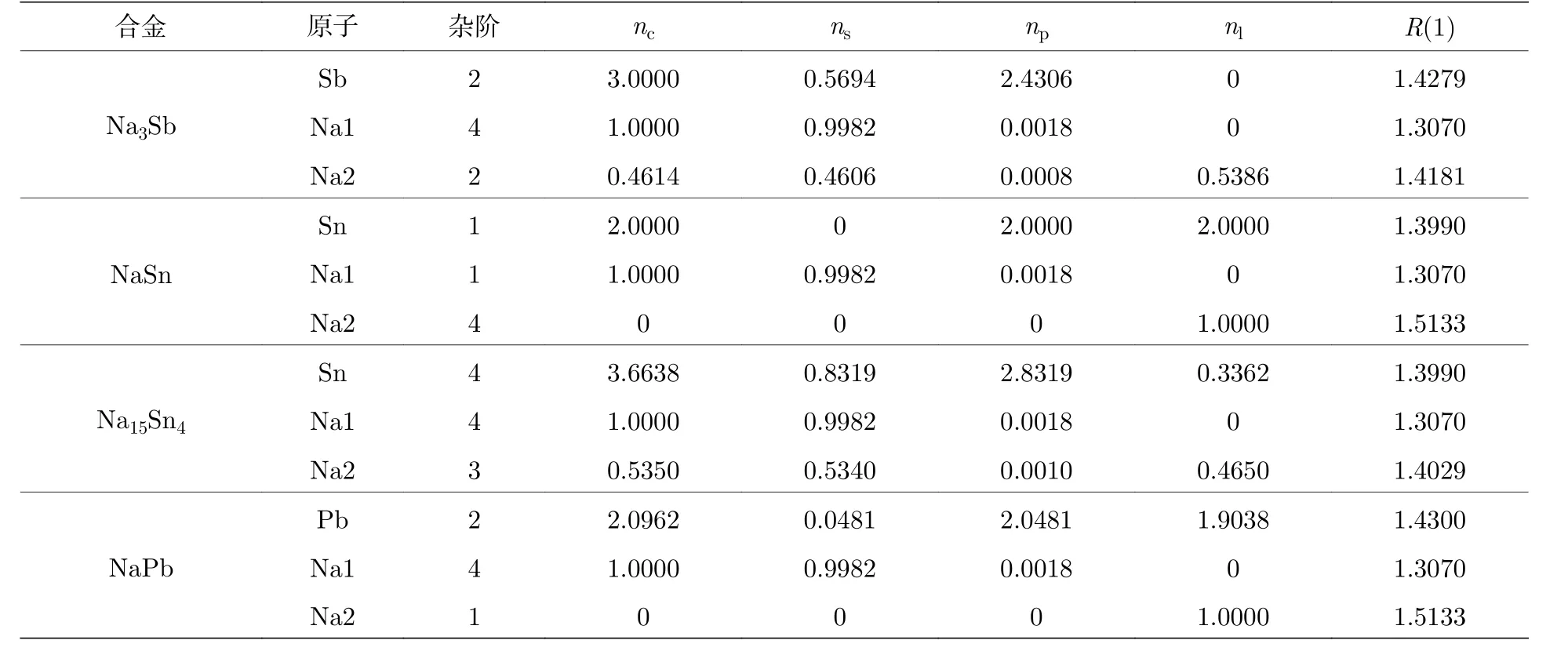
表6 阳极产物的价电子结构Table 6.Valence electron structures of anode products.

表7 正极合金的熔点、结合能与电势Table 7.Melting point, cohesive energy, and electric potentials of anode alloy.
4 结 论

表8 电池的开路电压Table 8.Open gate voltages of the battery.
应用固体与分子经验电子理论研究了Na||Sb-Pb-Sn 液态金属电池电极的价电子结构、熔点、结合能、电势及开路电压.研究表明: 阴极合金Na1-xIAx的熔点、结合能与电势都随着掺杂量x的增加而降低.理论的结合能与实验相符.其价电子结构研究揭示: 合金性能的变化与价电子结构密切相关, 随着掺杂量的升高, 晶格电子数nl明显降低, 这说明热声子优先破坏非成键的晶格电子在晶格空间的分布, 降低其电位能, 进而降低其对于阴极合金的熔点、结合能的贡献.对于阳极产物, 理论熔点与实验一致.由于其晶体结构复杂, 多种原子占位, 不同于结构简单的阴极合金.其性能不是与单独某种电子相关, 而是与共价电子和晶格电子相关联.开路电压与晶格电子密切相关, 与平均每原子的晶格电子数成反比, 平均晶格电子数最低的Na3Sb(0.2693)的开路电压最高, 而平均晶格电子最高的NaSn 的最低.对于Na||Sb-Pb-Sn 液态金属电池体系而言, 晶格电子扮演重要的角色, 可以调控电极的热、电性能.
附录A: 元素杂化表

表 A1 IA 族元素的乙种杂化表Table A1.B type hybrid table of IA group.
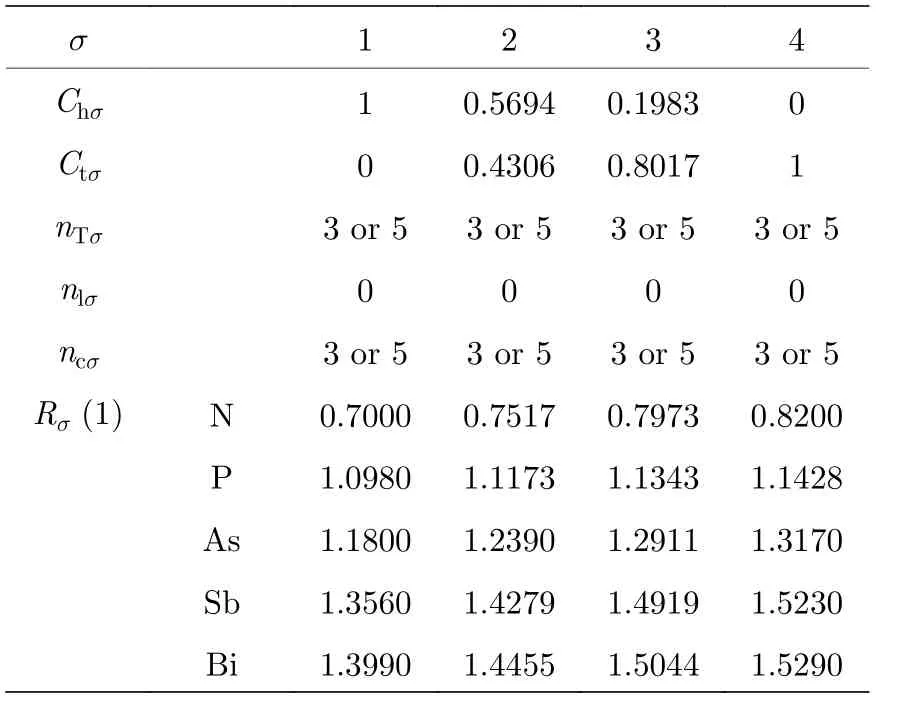
表 A2 VA 族元素的甲种杂化表Table A2.A type hybrid table of VA group.
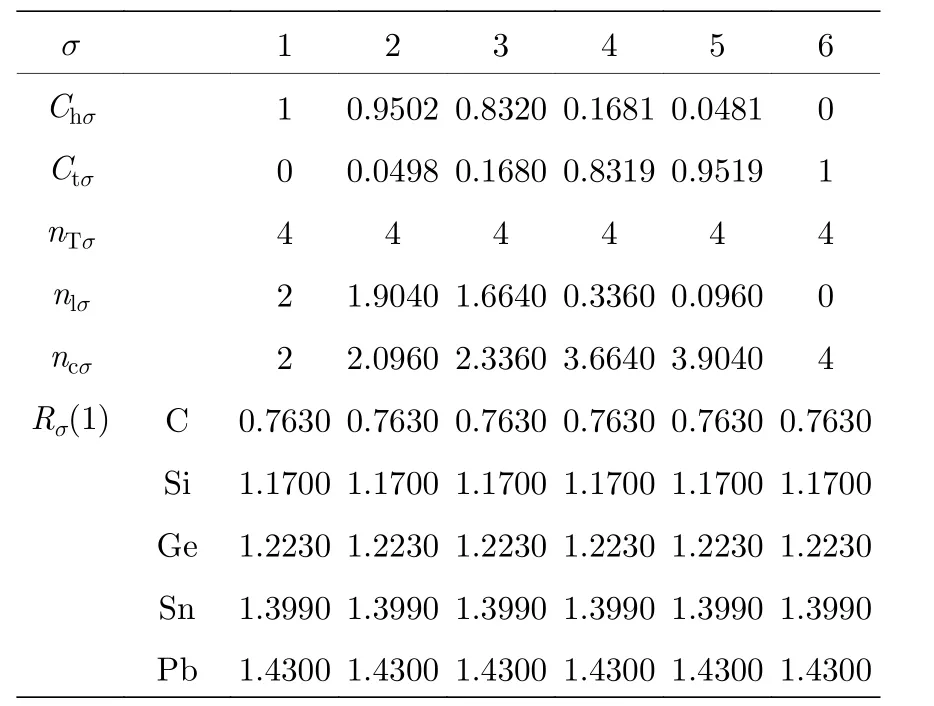
表 A3 IVA 族元素的甲种杂化表Table A3.A type hybrid table of IVA group.
