Mn-O共掺单层MoS2磁性和 光学性质的第一性原理研究
2021-04-08黄延彬魏节敏罗子江张振东
黄延彬, 丁 召, 魏节敏, 罗子江, 张振东, 郭 祥
(1.贵州大学大数据与信息工程学院, 贵阳 550025; 2. 贵州理工学院, 贵阳 550003; 3. 贵州财经大学信息学院, 贵阳 550025; 4. 贵州省微纳电子与软件技术重点实验室, 贵阳 550025; 5. 半导体功率器件可靠性教育部工程研究中心, 贵阳 550025)
1 引 言
自旋电子学是当前国内外研究的热点问题. 利用电子的自旋自由度可构造一些新的、更有效的逻辑器件以及新型的存储器件[1]. 当然,最重要的自旋电子特性体现在半金属铁磁性[2](half-metal ferromagnetism, HMFM)材料上,因其在自旋滤波器应用上有巨大潜力[3-4]. 目前研究结果表明氧化物[5]、硫化物[6]和石墨烯纳米棒[7]等材料中能实现HMFM,但鲜见在二维材料中的报道.
近年来,过渡金属二硫化物受到了人们的广泛关注[8].而二维MoS2材料是过渡金属二硫化物的典型代表,作为具有直接带隙(1.8 eV)的半导体,单层MoS2在电子和光子器件应用中具有优势[9-13]. Mo原子d轨道处于不饱和状态,因此可以在MoS2单层系统中通过掺杂其他元素调节其磁矩[14-16].二维MoS2具有优异的电学特性,例如室温下的自旋输运能力、几微米的自旋扩散长度, 这些特性使其成为自旋电子学上极具研究价值的材料[10-16].
Kong 等人[17]探究了O掺杂MoS2单层系统,发现该系统的带隙宽度随O掺杂浓度的增加而减小,并由掺杂前的直接带隙变为间接带隙. 曹娟等人[18]探究了V、Cr、Mn掺杂MoS2单层系统,发现相比于V和Cr掺杂,Mn掺杂的体系更加稳定,并且在室温下显示出稳定的铁磁性. Xie等人[19]探究了Mn-X(X=F、Cl、Br、At)共掺WS2单层系统,发现Mn掺杂和Mn-Br共掺杂WS2体系转变为具有完全(100%)自旋极化HMFM. 通过分析前期的相关研究发现,Mn-X共掺MoS2体系的研究相对较少. 因此,本文对单层MoS2进行Mn-O替位式掺杂,研究Mn-O共掺杂对MoS2单层体系能带结构、光学性质和磁性的影响.
2 计算方法和模型
本文的计算采用基于密度泛函理论(density functional theory, DFT)的剑桥系列总能量包(Cambridge serial total energy package,CASTEP)进行结构优化. 电子间相互作用的交换关联能通过广义梯度近似[20](general gradient approximation, GGA)和Perdew-Burke-Ernzerhof (PBE)函数进行处理. 平面波动态截止能量设置为470 eV,以确保计算的收敛性和高精度. 自洽能量的收敛精度为0.5×10- 5eV/atom,最大Hellmann-Feynman力在0.01 eV以内,最大离子位移在0.5×10-4nm之内,最大应力在0.02 GPa以内. 布里渊区域采用Monkorst-Park自动生成方案对全布里渊区积分K点进行取样,几何优化K点网格为5×5×1. 在所有计算中采用 4×4×1的MoS2单层超胞,晶格常数a=b=1.26 nm. 对于每个共掺杂结构,优化晶胞以获得最低总能量的晶格参数,对所有使用的模型采用周期性边界条件.
图1 单层MoS2的结构示意图Fig.1 Monolayer MoS2 structure diagrams
3 结果与讨论
3.1 结构分析
对未掺杂、O单掺、Mn单掺杂以及Mn-O共掺杂这四种MoS2单层系统体系优化后的结构参数如表1所示. 其中本征MoS2单层优化后,Mo-S键长为0.24 nm,S-Mo-S键角为81.962. O掺杂后,Mo-O键长为0.20 nm,比未掺杂的Mo-S键短. 由于VI-A族O原子的共价半径为0.73 nm,而S原子的共价半径为1.02 nm,共价半径小于S原子,因此共价半径小的O原子形成的Mo-O键短. Mn掺杂后,Mn-S键长为0.24 nm,小于Mo-S键长, S-Mn-S键角为82.759. 掺杂后键长的改变会产生局部应力,导致其键角也会随着键长的改变发生相应的变化,当键长变大时相对应键角会变小,反之变大,因此S-Mn-S键角略大于S-Mo-S. Mn-O共掺杂后,Mn-O键长为0.19 nm Mo-O-Mo键角为95.36.

表1 未掺杂、掺杂单层MoS2体系优化后的晶格常数
3.2 能带结构和磁性
图2为未掺杂、O和Mn单原子掺杂、Mn-O双原子共掺杂的单层MoS2体系能带结构,费米能级EF设定为零能量并由水平黑色虚线表示. 可以看到,本征MoS2单层为直接带隙,禁带宽度1.738 eV,略低于实验值1.740 eV[21-23],这是因为基于密度泛函理论计算的方法会低估带隙值的大小[24],未掺杂的单层MoS2系统因其自旋向上和自旋向下通道中的能带被完全对称地填充,因此呈现出非磁性半导体特性. 当O单原子掺杂MoS2单层系统后,MoS2仍然表现为非磁性半导体特性,但是能带结构由直接带隙转变为间接带隙,禁带宽度转变为1.624 eV. Mn掺杂和Mn-O共掺杂之后,由于自旋向上通道中少许杂质带与费米能级交叠,导致掺杂系统出现金属特征,而自旋向下通道仍保持半导体特征,但具有较小的自旋间隙,分别为0.158和0.396 eV,表明Mn掺杂和Mn-O共掺杂单层MoS2系统变为半金属(half metal, HM)铁磁体,具有完全(100%)自旋极化.图2(b)和2(c)表示自旋向上通道,图2(e)和2(f)表示自旋向下通道. Mn掺杂后,体系磁矩为1μB,与Xie等人[19]计算结果吻合,其中S贡献-0.6μB,Mo贡献-0.69μB,Mn贡献2.29μB. Mn-O共掺杂后,体系磁矩为1.08μB,其中O贡献0.01μB,S贡献 -0.06μB,Mo贡献 -0.02μB,Mn贡献1.15μB. Mn掺杂体系磁矩主要来源于Mn-3d态的自旋不对称.

图2 未掺杂、Mn掺杂、O掺杂和Mn-O共掺杂MoS2单层系统的能带结构图
3.3 态密度
了解电子态密度(density of states, DOS)是分析材料的电学和光学性质的前提.图3中绘制了总态密度(total density of states, TDOS)和投射到掺杂原子Mn-3d、Mo-4d、S-3p和O-2p上的分态密度(partial density of states, PDOS). 在Mn单掺杂和Mn-O共掺杂的单层MoS2系统,正值和负值分别代表自旋向上和自旋向下通道,垂直红线表示费米能级EF并设定为零能量.图3(a)显示在纯单层MoS2系统中,自旋向上和自旋向下的态密度是完全对称的,因此展现出非磁性半导体特性.图3(c)和3(d)所示的总态密度TDOS说明在Mn掺杂和Mn-O共掺杂单层MoS2系统中,部分占据的杂质态仅在自旋向上通道中出现,说明在该费米能级具有完全(100%)自旋极化的HM铁磁特性. 相应的较低部分PDOS表明部分占据杂质态主要由较近Mo-5d态杂化的Mn-3d态贡献.
Mn单掺杂和Mn-O共掺杂MoS2单层系统由于电子之间的交换作用,在费米能级EF附近,自旋向上和自旋向下的态密度发生了自旋劈裂,且自旋向上与自旋向下的电子数目不等,在3d能带中形成未被抵消的自发磁矩,因而发生自发磁化. Mn掺杂的MoS2单层体系在靠近价带顶(CBM)的位置出现了明显的杂质能级并有能级劈裂现象,其在自旋向下的通道中穿过费米面,杂质能级来源于Mo-4d和Mn-3d轨道,表明体系的磁矩主要局域在 Mo和 Mn上. Mn掺杂的MoS2单层体系在靠近导带底的位置出现了明显的杂质能级,其主要由 Mo-4d和Mn-3d轨道电子组成. 靠近价带顶的位置发生了自旋劈裂,自旋向上的部分穿过费米面,自旋向下的部分依然位于费米能级以下,表现出半金属性,其主要由 Mo-4d和Mn-3d轨道电子组成,这表明 Mn掺杂的MoS2单层体系的磁矩主要局域在Mo和 Mn上. 从 态密度上可以看出,在Mn掺杂的MoS2单层体系中,在费米面附近Mn-3d、Mo-4d和S-3p态之间发生了强烈的耦合作用,说明Mn是引起体系磁性的主要原因.

图3 未掺杂、Mn掺杂、O掺杂和Mn-O共掺杂MoS2单层系统的总态密度TDOS与投射到掺杂原子Mn-3d、Mo-4d、S-3p和O-2p上的分态密度PDOS(垂直虚线代表费米能级EF为零值)
3.4 光学性质
为探究Mn-O共掺单层MoS2系统光学性质的影响,在图4(a)和4(b)中分别绘制介电常数的实部ε1(w)和虚部ε2(w). 对于电磁辐射,介电常数ε(w)可用作描述系统的线性响应,通常定义为:
ε(w)=ε1(w)+ε2(w)
(1)
其中实部ε1(w)反映了介质的介电特性, 而虚部ε2(w)反映了半导体/绝缘体复合材料对电磁波的吸收特性.图4(a)计算出未掺杂,Mn单掺杂,O单掺杂,Mn-O共掺的静态介电常数ε1(0)分别为:6.125、 8.8415、 6.105、 6.576,我们发现Mn掺杂和Mn-O共掺杂后实部介电函数ε1(w)的静态值高于未掺杂MoS2单层系统,并且Mn掺杂后的静态值为四个对比组的最大值达8.841 5,说明Mn掺杂MoS2单层体系具有较好的介电特性. 随着光子能量的增加,实部ε1(w)的值呈先增加后减小再增加的趋势,峰值分别为:8.613、 8.510、7.580、7.972,对应的光子能量分别为:2.049、2.001、2.026、1.923 eV,介电常数在光子能量5.265~7.697 eV区间变为负值,当ε1(w)>0时,光子在材料中传播,对于ε1(w)<0,电磁波被衰减,对于ε1(w)=0,只可能存在纵向极化波.图4(b)显示随着光子能量的增加,介电函数的虚部ε2(w)也在增加,在光子能量2.9 eV附近ε2(w)达到最大值. 在0~0.9 eV范围内,由3.3中所述铁磁特性(HM)导致Mn掺杂MoS2系统的介电函数ε(w)值随着光子能量的增加而减小.
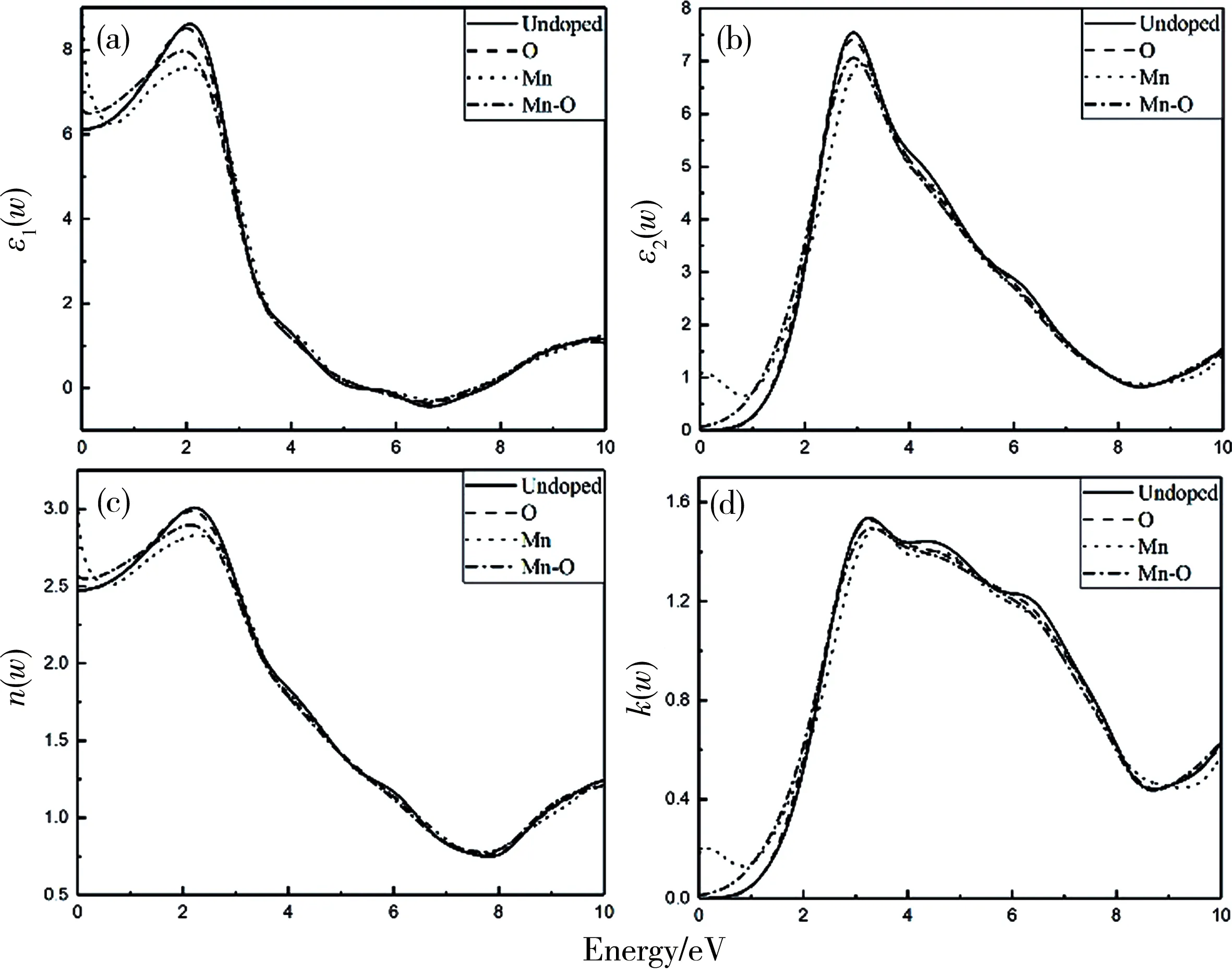
图4 (a)和(b)分别为未掺杂、Mn掺杂、O掺杂和Mn-O共掺杂MoS2单层系统的介电常数的实部ε1(w)和虚部ε2(w);(c)和(d)分别为折射系数的实部n(w)和虚部k(w)
在图4(c)和4(d)分别绘制折射系数的实部n(w)和k(w)随光子能量变化的曲线,发现他们与介电常数的实部ε1(w)与虚部ε2(w)的图形相似. 其中未掺杂、Mn单掺杂、O单掺杂和Mn-O共掺杂MoS2单层系统的静态折射系数n(0)分别为3.007、 2.985、 2.829、 2.896. 在图4(d)中0~4 eV低能量区域,四个对比组k(w)的值随着光子能量的增加而增加,直至最大值,分别为1.536、 1.527、1.494和1.494,同时在3.0~7.5 eV区域内表现出振荡行为. 与其他三种掺杂情况不同的是,由于Mn掺杂MoS2单层系统出现了铁磁性质,
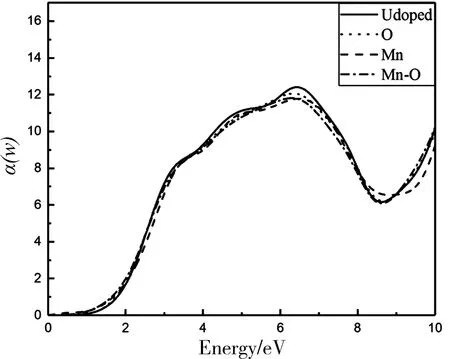
图5 未掺杂、Mn掺杂、O掺杂、Mn-O共掺杂MoS2单层系统的吸收系数α(w)
导致其k(w)值在0.2 ~0.83 eV范围内随着光子能量的增加而减小,在0.83 eV之后随着光子能量的增加而增加,并在3.422 eV达到最大值1.494.
图5为不同掺杂条件下单层MoS2的光吸收谱,在红外光波段(780 nm ~ 1 mm),它们的吸收边分别为0.83、0.126、0.869和0.556 eV,对应的波长分别为1 495.80、 9 853.29、 1 428.67、 2 232.94 nm,这说明MoS2单层系统掺杂后出现一定的红移现象,在红外光探测上表现出良好的特性. 未掺杂与掺杂后的吸收系数峰值分别为12.407、 11.783、 12.064和11.813,对应的能量分别为6.442、 6.484、 6.335和6.295 eV. 在低能量区域(0~2.5 eV),掺杂后吸收系数较未掺杂的有所增强,其中Mn-O共掺杂的吸收增强最为显著.
4 结 论
本文基于第一性原理自旋极化密度泛函计算了本征单层MoS2、O、Mn和Mn-O掺杂MoS2单层系统电子结构、磁性和光学性质. 发现掺杂后的MoS2单层系统的晶胞参数和电子结构发生了一定变化,本征MoS2单层系统的禁带宽度为1.738 eV,为直接带隙非磁性半导体. O单原子掺杂MoS2单层系统后转变为禁带宽度为1.624 eV的间接带隙非磁性半导体. 在Mn和Mn-O掺杂后,MoS2单层系统从自旋向上和自旋向下完全对称的非磁性半导体转变为拥有磁矩为1μB和1.08μB的铁磁半导体. 同时发现Mn-O共掺杂后,在低能量区域,MoS2单层系统在光学性质上相比未掺杂和O单掺杂的MoS2系统有所增强,并出现红移现象.
