基于FASTmrEMMA、最小角回归和随机森林的全基因组选择新算法
2021-03-29孙嘉利吴清太温阳俊张瑾
孙嘉利,吴清太,温阳俊,张瑾
(南京农业大学理学院,江苏 南京 210095)
许多动植物的重要性状和人类的复杂疾病都是数量性状,检测、发掘与数量性状关联的基因并进行全基因组选择,对动植物的性状分析及人类疾病的预防和治疗都具有重要意义[1-5]。全基因组选择(genomic selection,GS)又称全基因组预测(genomic prediction,GP),它是Meuwissen等[6]提出的一种利用全基因组高密度分子标记数据及原始训练群体表型数据建立预测模型,估计每个分子标记遗传效应,从而预测个体全基因组育种值(genomic breeding value,GBV)的方法。由于GS无需鉴定与目标性状显著关联的位点,即使微效标记也都能被捕获,只需个体遗传信息即可计算其育种值,这大大缩短了育种周期,目前已广泛应用于动植物的全基因组选择[7-10]。
近年来,研究人员陆续提出了许多GS方法,例如,应用广泛的最佳线性无偏预测(best linear unbiased prediction,BLUP),它是基于系谱信息来定义个体间亲缘关系矩阵,校正环境和非随机交配造成的偏差,从而提供个体育种值的无偏估计值。GBLUP(genomic BLUP)[11]是利用全基因组遗传标记计算遗传关系矩阵来预测表型缺失值。RR-BLUP(ridge regression BLUP)[6]是利用压缩估计获得标记效应来进行全基因组预测,但可能会被过度压缩。BayesA和BayesB[6]可以有效弥补RR-BLUP的不足,它们假设每个标记效应服从先验分布,其中BayesB的先验分布为混合分布。此外,还有BayesC[12]、Bayesian LASSO[13]等方法。上述GS大多基于压缩估计方法,其计算速度会随着数据维度的增加而受到限制,不适用于现代测序技术产生的海量遗传标记数据,且尚未考虑群体结构和多基因背景,从而影响全基因组预测的准确度。
植物交配设计相对复杂,考虑群体结构和多基因背景可以有效提高全基因组预测的准确度。TSLRF[14]是一种结合了最小角回归(least angle regression,LARS)[15]和随机森林(random forest,RF)[16]方法的两阶段关联分析算法。它首先应用FASTmrEMMA方法[17]进行群体结构和多基因背景控制,再通过LARS方法选择与目标性状潜在关联的单核苷酸多态(single nucleotide polymorphism,SNP),并用此SNP子集建立RF模型预测个体的GBV,即全基因组预测值。本研究以拟南芥自然群体的模拟数据和真实数据为研究对象,利用TSLRF方法针对植物数量性状进行全基因组预测,其结果与TSRF[18]、RF和GBLUP进行比较,验证TSLRF方法对植物数量性状全基因组预测的准确性和可行性,为后续的分子育种和优异亲本组合预测提供理论依据和现实参考。
1 材料与方法
1.1 模拟试验材料
以拟南芥数据[19]为模拟试验材料。按照文献[20]的方法选取其中的10 000个SNP标记,它们分别来自于5条染色体片段,其位置分别介于染色体1至5的11 226 256~12 038 776 bp、5 045 828~6 412 875 bp、1 916 588~3 196 442 bp、2 232 796~3 143 893 bp和19 999 868~21 039 406 bp[20]。选取6个QTN(quantitative trait nucleotide),分别设置在染色体1的11 298 364、11 655 607 bp标记上(遗传率设置为10%、5%)和染色体2的5 066 968、5 134 228、5 464 675、6 137 189 bp标记上(遗传率设置为5%、15%、5%、5%),其等位基因频率均为0.30;总体均值和残差均设置为10.0,样本容量为199,随机生成1 000次重复表型数据。该模拟数据集已应用于多项研究[17,20-21]。
为考察不同表型缺失率的预测效果,针对上述模拟数据集的199个个体,分别以5%、10%、15%和20%的缺失率随机删除个体表型数据,构造缺失数据集,用来比较TSLRF、TSRF、RF和GBLUP这4种方法在不同缺失率下的全基因组预测准确度(MAE和MSE)、预测模型拟合度和计算时间。
1.2 真实试验材料
真实数据集[19]来自拟南芥自然群体的199个个体,216 130个SNP(http://www.arabidopsis.usc.edu/),考虑长日照花期(LD)、春化长日照花期(LDV)和短日照花期(SD)3个花期相关性状(https://www.arabidopsis.org/portals/genAnnotation/index.jsp),其表型缺失率分别为16.0%、15.6%和18.6%。利用TSLRF、TSRF、RF和GBLUP预测表型值,并利用预测值和已有观测值进行全基因组关联分析,挖掘与性状关联的基因。
1.3 TSLRF方法
TSLRF先将模型中的群体结构Q矩阵设为固定效应来校正群体结构效应,并借助FASTmrEMMA[17]将个体间多基因背景和误差变异转换成标准正态离差来校正多基因背景,便于后续进行变量选择;然后用LARS选择出最可能与目标性状关联的SNP子集;再用此SNP子集建立随机森林模型预测个体的GBV。其过程如图1所示。

图1 基于最小角回归和随机森林的两阶段算法 (TSLRF)的流程图Fig.1 The flow chart of two-stage algorithm based on least angle regression and random forest(TSLRF) LARS:最小角回归Least angle regression;SNP:单核苷酸多态Single nucleotide polymorphism;RF:随机森林Random forest;GBV:全基因组育种值Genomic breeding value.
1.3.1 遗传模型遗传模型如下:
y=1μ+Wα+Zγ+u+ε
(1)
式中:y=(y1,…,yn)T为自然群体的表型向量,n为样本容量;1表示分量为1的n×1向量;μ为总体均值;α表示固定的群体结构效应;γ表示p×1的标记随机效应向量,p是标记数量;W和Z分别是α和γ对应的设计矩阵;u表示n×1的多基因随机效应向量;ε为n×1的误差向量。

(2)


(3)

yc=1μ+Wcα+Zcγ+εc
(4)
式中:yc=Cy;Wc=CW;Zc=CZ;εc=Cu+Cε~MVNn(0,σ2In)。
Y=Xβ+εc
(5)
式中:Y为校正后的表型值;X为标准化后的基因型;β为包含固定效应和随机效应的向量;εc为服从MVNn(0,σ2In)的误差向量。
1.3.3 LARS算法采用LARS算法选择出与表型Y具有绝对相关最大的变量,如xj1,则当前路径为s1*xj1,其中s1是xj1为与当前残差之间相关系数的符号。按照这一路径前进,直到出现另一变量与当前残差的相关系数更大,记为xj2,将其加入回归模型中并重复上述过程。设置LARS算法迭代到第k步,选择出k个与目标性状潜在关联的SNP来构建RF模型。
1.3.4 RF算法针对LARS选择出的变量集{xi1,xi2,…,xik}(i=1,2,…,n)以及原始表型观测值,RF先使用Bagging算法生成500个自采样样本数据集,每次未被抽到的样本组成500个袋外(out of bag,OOB)样本数据集;然后通过节点随机分裂技术,从每棵树的每个节点处的k个变量中随机抽取mtry个变量作为备选分支变量(回归树一般取mtry=k/3),按照节点分裂原则,选择最优分支进行分裂。生成500棵CART树构建RF模型,并预测所有个体的全基因组育种值。其中,mtry选取能使RF模型获得最佳模型拟合度的参数值。本文中,RF将所有CART树的平均值输出作为预测值。
TSLRF可通过R软件实现,其中LARS和RF可分别通过程序包lars和randomForest实现。
1.4 评估指标
采用平均绝对误差(MAE)和均方误差(MSE)来评估预测准确度,用皮尔森相关系数(r)评估预测模型的拟合度。MAE计算公式为:
(6)

(7)
与MAE相比,MSE取误差值的平方,增大了误差的作用,即对误差的估计更加敏感,是预测效果评估中具有代表性的指标。MSE越大表示准确度越低,越小表示准确度越高。
皮尔森相关系数(r)计算公式为:
(8)

2 结果与分析
2.1 模拟数据的全基因组选择
为了验证TSLRF的全基因组预测能力,利用TSLRF、TSRF、RF和GBLUP对不同表型缺失率(5%、10%、15%和20%)的模拟数据集进行分析。这4种方法都是基于R软件实现的,其中,3种随机森林方法(TSLRF、TSRF和RF)通过程序包randomForest实现;GBLUP通过程序包GAPIT实现。
计算1 000次重复的全基因组预测值与观测值之间的MAE和MSE。结果表明,随着表型缺失率的增大,4种方法的MAE(图2-A)和MSE(图2-B)增大。这意味着随着表型缺失率的增大,全基因组预测准确度不断降低,并呈现出越来越显著的差异,这与事实相符。在4种缺失率下,TSLRF的MAE显著小于其他方法(方差分析检验,P=1.28×10-6),其次为TSRF和RF;GBLUP的MAE最大,在缺失率为5%和10%的情况下,大约是TSLRF的2倍。这说明TSLRF预测值与实际观测值间的MAE最小,即预测准确度最高。4种方法MSE的总体趋势与MAE的情况相似。在4种缺失率下,RF的r最大,位于0.88~0.95;TSLRF在5%、10%、15%表型缺失率下仅次于RF,位于0.90~0.94,在20%缺失率时,TSLRF(0.89)要稍优于RF(0.88);而TSRF和GBLUP的全基因组预测能力要低于RF和TSLRF,TSRF的r位于0.85~0.91,GBLUP的r最小,位于0.84~0.91(图2-C)。总体来说,RF的预测值与观测值之间的皮尔森相关系数最大,TSLRF仅次于RF,而TSRF和GBLUP的模型拟合度不能令人满意。

图2 不同表型缺失率下TSLRF、TSRF、RF和GBLUP全基因组选择的平均绝对误差(A)、 均方误差(B)和皮尔森相关系数(C)(重复1 000次)Fig.2 The mean absolute error(A),mean squared error(B)and Pearson correlation coefficient(C)of TSLRF,TSRF, RF and GBLUP in the genome-wide selection under various phenotypic missing rates(1 000 repeats) TSLRF:基于最小角回归和随机森林的两阶段算法Two-stage algorithm based on least angle regression and random forest;TSRF:基于随机森林的两阶段变量选择Two-stage stepwise variable selection based on random forest;RF:随机森林Random forest;GBLUP:全基因组最佳线性无偏预测Genomic best linear unbiased prediction.
由表1可见:随着表型缺失率的增大,4种方法的计算时间逐渐降低。在相同表型缺失率下,TSLRF和RF的计算时间均少于100 min,其中RF的计算时间最短,TSLRF仅次于RF,这是由于TSLRF进行全基因组的群体结构和多基因背景控制需要花费一定的时间;TSRF的计算时间约为200 min;GBLUP的计算时间最长,约为400 min。

表1 不同表型缺失率下1 000次模拟数据集的TSLRF、TSRF、RF和GBLUP计算时间
2.2 真实数据的全基因组选择
真实拟南芥数据包括199个个体,216 130个SNP,3个与花期相关的LD、LDV和SD性状,其表型缺失率分别为16.0%、15.6%和18.6%。
采用TSLRF、TSRF、RF和GBLUP方法预测表型值,利用表型预测值和观测值进行关联分析,得到显著SNP并挖掘基因,考察这4种方法的全基因组预测能力。分别将TSLRF、TSRF和RF方法中重要性排名前20及GBLUP方法中效应估计值排名前20的SNP视为QTN。利用TAIR基因库(https://www.arabidopsis.org)查找位于QTN附近20 kb的关联基因。从表2可见:TSLRF、GBLUP、TSRF、RF预测到被证实的基因数分别为40、30、24和21个。TSLRF基因预测能力最强,GBLUP次之。

表2 4种方法预测的拟南芥LD、LDV和SD性状前20个显著QTN附近的已知基因数
TSLRF预测出与同一性状显著关联的多个基因簇(表3),如, 位于染色体1的2 005 921 bp标记附近的基因AT1G06515和AT1G06520被预测到与LD相关;位于染色体5的26 004 094 bp标记附近的基因AT5G65050、AT5G65060、AT5G65070和AT5G65080被预

表3 TSLRF预测的拟南芥LD、LDV和SD性状前20个显著QTN附近的已知基因Table 3 The known genes in the vicinity of the top 20 significant QTN for each of the LD,LDV and SD traits in A.thaliana using the TSLRF method
测到与LDV相关。TSLRF预测到的一些显著关联的基因,同时被其他方法预测到,例如,与LDV显著相关的位于染色体2的9 965 680、9 964 693、9 966 813和9 967 491 bp标记附近的基因AT2G23380可被TSLRF和GBLUP预测到;与SD显著相关的位于染色体4的13 996 527、13 997 089和13 998 150 bp标记附近的基因AT4G28190和AT4G28280可被TSLRF和TSRF同时预测到。由此可见,与其他3种方法相比,TSLRF具有更有效、更准确的基因预测能力。
4种方法对真实数据分析的计算时间如表4所示。结果表明,TSLRF和RF的计算时间最短,均小于2 min,这与模拟研究的结果一致;GBLUP的计算时间约为25 min;TSRF的计算时间最长,至少需要60 min。因此,从运行效率来看,利用TSLRF进行全基因组预测是可行的。
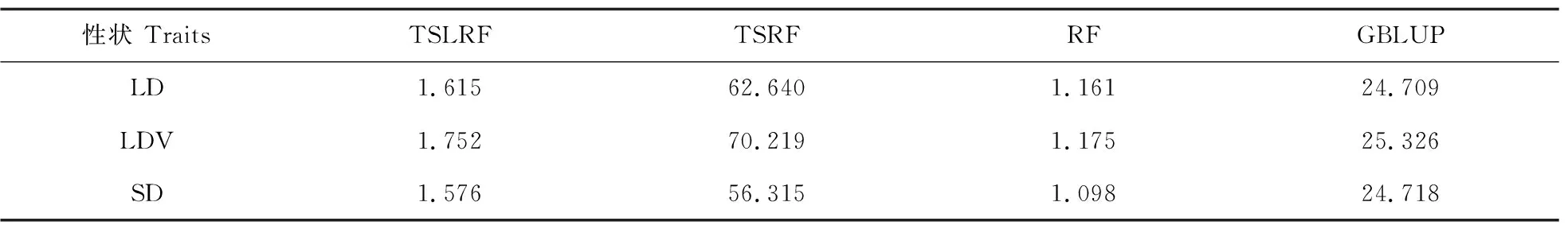
表4 利用4种全基因组预测拟南芥LD、LDV和SD性状的计算时间Table 4 Computing times for the LD,LDV and SD traits in A.thaliana using 4 methods min
3 结论与讨论
针对动植物重要性状和人类复杂疾病进行全基因组预测,对作物改良和疾病防治具有重要意义。本文利用TSLRF方法进行全基因组预测,其先校正群体结构和多基因背景,再结合变量选择方法LARS和机器学习方法RF进行预测,既能显著提高模型准确度与拟合度,又能显著提升全基因组的预测能力与效率。
模拟研究和真实数据分析均表明,表型缺失率是影响预测准确度和后续关联分析检测能力的重要原因之一。随表型缺失率增大,计算时间减少,预测值与观测值之间的MAE和MSE增大,全基因组预测准确度显著降低,模型拟合度也降低。真实数据分析中,根据随机森林相关研究[22-23],本文以经验排名选择显著SNP个数,分别选取前20、30、50的SNP。排名20以后(21~50)的SNP重要性得分明显下降,与非关联SNP之间差距不显著;且被TAIR基因库证实的基因数也未显著增加。因此,本文选取排名前20的SNP作为显著关联标记。
从全基因组预测运行速度来看,TSLRF和RF比其他2种方法(TSRF和GBLUP)快。GBLUP在计算遗传关系矩阵上需要花费一定的时间,因此它的总计算时间最长;TSRF由于重复采样、多次计算重要性得分,计算时间也较长,大约是TSLRF的60倍。综上所述,结合全基因组预测的准确度、拟合度以及计算时间等指标,TSLRF是高效、可行的全基因组选择方法,这为机器学习方法应用于海量数据的全基因组预测和全基因组关联分析提供了新的途径。
