脊髓毛细胞型星形细胞瘤5例临床分析
2021-02-26赵斌钟鹏马强肖华亮
赵斌,钟鹏,马强,肖华亮
毛细胞型星形细胞瘤(pilocytic astrocytoma,PA)是Penfield于1937年根据肿瘤细胞两端突起为细长的毛发样胶质纤维而命名的颅内肿瘤;在2007版WHO中枢神经系统肿瘤分类中为WHO Ⅰ级[1]。PA是一种生长缓慢、边界清楚,常出现囊性变,好发于儿童及青少年的星形胶质细胞瘤,多数发生在20岁以前,成年及老年人少见,无明显性别差异。发生于脊髓的PA少见,包括有原发性和继发性脊髓PA,继发性PA由颅内PA通过脑脊液扩散引起[2-4]。陆军特色医学中心2016年1月—2019年12月收治5例脊髓PA患者;本研究对患者的临床资料进行回顾分析,以探讨PA的临床、影像学、病理学特点及治疗与预后。
1 临床资料
1.1 临床表现 本组患者中男4例,女1例;年龄24~66岁,平均年龄44.6岁;病程6~180 d,中位数20 d。首发症状以肢体麻木、疼痛及颈、腰部疼痛为主,部分患者的症状进行性加重。其中4例患者出现肢体或颈、腰部疼痛,2例患者出现肢体麻木,1例患者出现右侧上下肢体无力,1例患者出现四肢无力。脊髓功能状态用McCormick 分级标准评价[5],McCormick 分级Ⅰ级者3例、Ⅱ级1例、Ⅳ级1例。见表1。
1.2 影像学检查 5例患者术前均行MRI检查(平扫+增强扫描)。5例患者的肿瘤均位于脊髓内,T1WI为低信号,T2WI为高信号;病变位于颈段者2例、胸段1例、腰段1例、颈至胸段1例,病变累及1个至多个节段;肿瘤边界清楚或较清楚者4例,边界欠清1例;其中1例患者病变累及C1至T7并伴有脊髓空洞,1例患者出现周围脊髓水肿。增强扫描示病变呈明显强化者2例,片状强化或不均匀强化者3例。见表2、图2。

表1 本组患者的一般资料、病变部位、首发症状及McCormick 分级

表2 本组患者脊髓PA的MRI表现
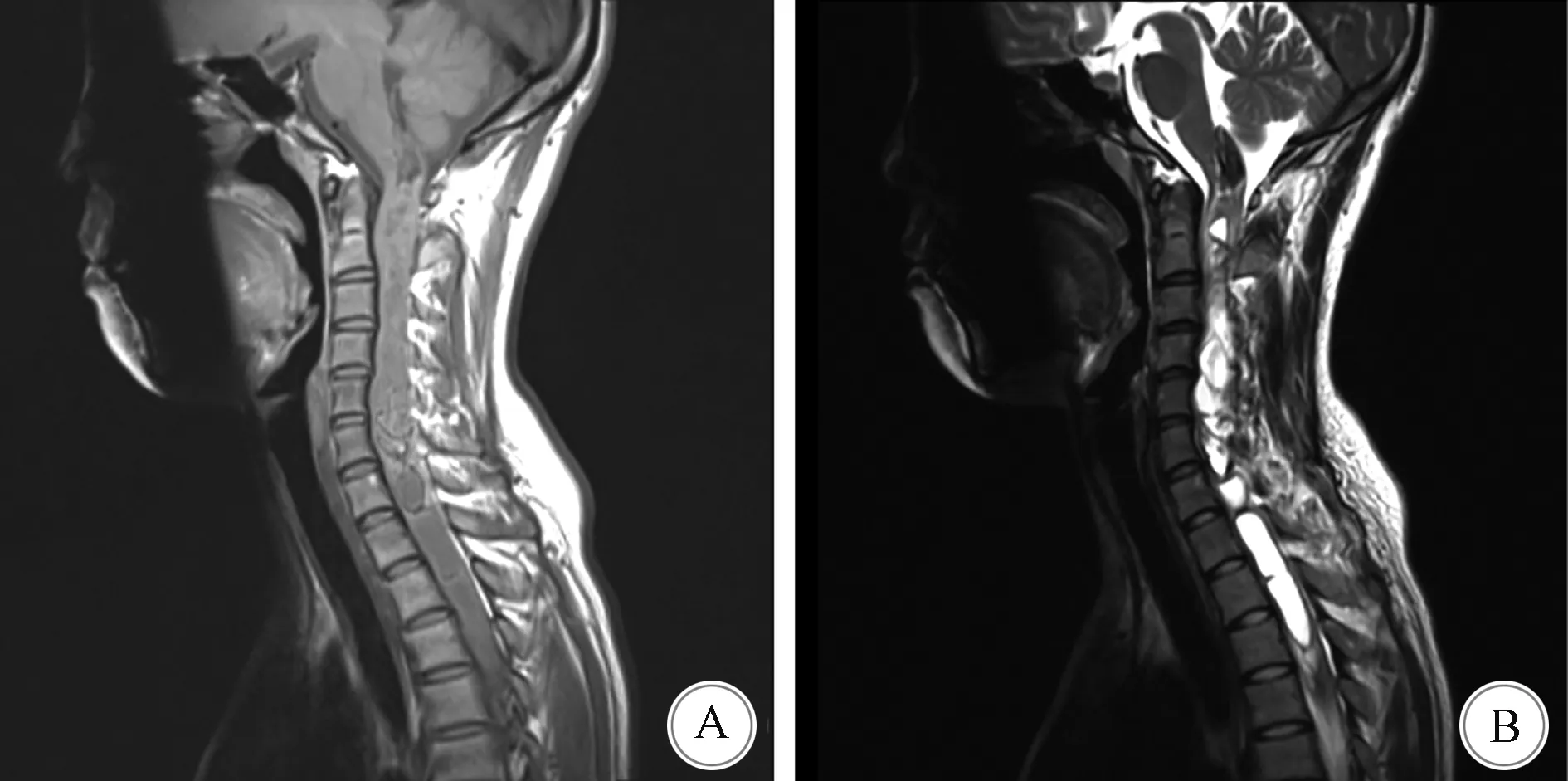
A:MRI T1WI示C1-T7等信号病灶; B:T2WI示病灶为高信号,边界清楚,伴脊髓空洞形成图1 影像学检查结果
1.3 病理学检查 肿瘤标本HE染色,光镜下观察显示,2例患者出现典型Rosenthal纤维及嗜酸性小体,另3例患者无此改变,但有毛细胞形态特点;5例患者均出现血管增生伴扩张出血,2例患者血管壁玻璃样变,其中1例患者血管壁玻璃样变较广泛;肿瘤伴随改变:囊性变者1例,黏液背景2例,灶性钙化2例,细胞退变1例,煎蛋样细胞1例,类似少突胶质瘤区域。免疫组化染色显示:GFAP、Vimentin、S-100均阳性,Nestin、CD56、olig-2部分病例出现灶性阳性,ATRX未缺失,IDH-1、MGMT、EMA、PR、P53及CyclinD1均为阴性,Ki-67约1%~2%。5例患者的病理诊断均为脊髓PA(WHO Ⅰ级)。
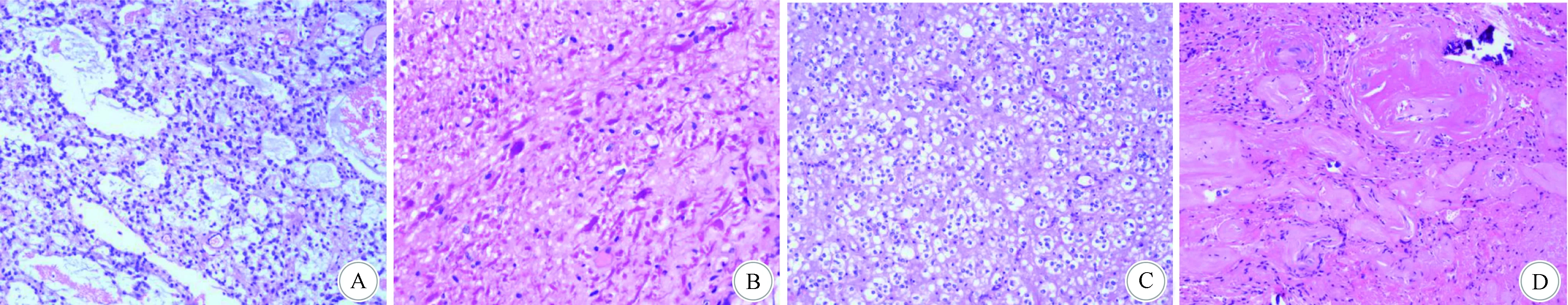
A:显示疏松微囊区; B:显示致密区中的Rosenthal纤维; C:显示部分呈少突样区域; D:显示血管增生伴玻璃样变性及钙化灶图2 组织病理学检查结果(HE染色,×200)
1.4 治疗与预后 5例患者均行手术全切除肿瘤,术后均行患者辅助康复治疗。术后随访观察6~36个月,其中4例患者症状明显好转,1例患者症状改善并无加重,继续康复中。
2 讨 论
2.1 脊髓PA的临床特点 PA临床较少见,据国内文献报道其约占颅内肿瘤的1.6%[6]。PA可发生于神经系统任何部位,以小脑最常见,位于小脑者占80%[7];脊髓PA少见,发生在脊髓的PA仅占0.17%[6]。PA多发于儿童及青少年,约75%~88%的患者发病时小于20岁;脊髓PA的发病率极低,但在儿童和青少年中,其约占所有髓内胶质瘤的21%[8]。PA是儿童最常见的脊髓肿瘤,在成年人脊髓肿瘤中也仅次于室管膜瘤排在第2位[9]。本组5例患者均为成年人,且均大于20岁。脊髓PA的临床表现为肿瘤膨胀性生长引起的非特异性症状,主要为肢体疼痛、运动障碍,可伴有肢体麻木等症状。首发症状轻重不一,可逐渐加重;根据肿瘤累及的脊髓节段,重者可同时出现四肢运动障碍。本组5例患者的首发症状都比较轻,为颈部、腰部或四肢疼痛或伴麻木;2例患者症状逐渐加重,其中1例患者出现一侧肢体运动及感觉障碍,1例患者出现四肢运动障碍及感觉障碍,这2例患者的病变累及节段分别是C4-C5和C1-T7。
2.2 影像学表现 MRI是脊髓PA的最佳检查方法。脊髓的PA好发于胸段,其次是颈段,再次是腰段[9];但也有个案报道累及全脊髓的PA[10]。本组患者中发生于颈椎者2例、胸椎1例、腰椎1例、颈至胸椎1例。PA在MRI上表现为实性或囊实性肿块,可累及多个脊髓节段,脊髓明显增粗,与正常脊髓分界可清亦可不清楚[11];肿块在T1WI多表现为等低信号,T2WI为高信号或稍高信号;伴有脊髓空洞的表现信号混杂(图1)。增强扫描示PA实性部分多有明显强化,囊变、坏死区域无强化。脊髓PA增强扫描的强化程度不及颅内PA明显,但也可呈轻、中、重不同程度的强化,多数呈絮状强化[10];但无强化的PA也不少见[12]。髓内PA易被MRI诊断为室管膜瘤或血管母细胞瘤,室管膜瘤实体部分血供丰富,有出血倾向,常有不同程度的含铁血黄素沉积,出血区呈低信号,在T2WI更为明显,增强扫描实体部分强化明显。PA在影像学上缺乏特征性表现,因此术前诊断髓内PA较困难。
2.3 病理学特点 PA的瘤细胞密度较低,呈双相性结构,有Rosenthal纤维的致密双极性细胞区,同时具有微囊和嗜酸性小体的多极性疏松区。Rosenthal纤维HE染色呈圆形、椭圆形或不规则形的半透明红染物,排列与胶质纤维一致,多少不等。嗜酸性小体是位于胞浆内的嗜伊红球形物质,PAS染色阳性,α-抗胰蛋白酶阳性。Rosenthal纤维常聚集在致密区,嗜酸性小体主要见于微囊组织;Rosenthal纤维与嗜酸性小体对PA诊断有提示作用,但并非诊断必须,也不是PA所特有。几乎所有PA可见不同程度的血管增生,有似血管瘤样区域,也有肾小球样血管区域,并可伴出血;多数PA出现增生血管管壁增厚、玻璃样变性(图2)。PA瘤细胞可出现异型性,但大多数病例没有或少见核分裂,血管的增生及瘤细胞的异型性并不是恶变的特征。部分病例可出现类似少突胶质细胞瘤样煎蛋样细胞,但细胞密度低。本组患者中,有2例患者出现典型Rosenthal纤维与嗜酸性小体,另3例患者无此改变,但有毛细胞形态特点;5例患者均出现血管增生伴扩张出血,其中2例患者血管壁玻璃样变(1例患者血管玻璃样变较广泛);此外,1例患者伴有囊性变,2例患者伴有黏液背景,2例患者伴灶性钙化,1例患者伴细胞退变,1例患者伴有煎蛋样细胞,类似少突胶质瘤区域。
2.4 免疫组化及分子遗传学特点 PA在免疫组化上并无特异性表达,免疫组化表达显示为胶质源性分化的肿瘤,GFAP、Vimentin、S-100呈阳性表达,Nestin、CD56、olig-2部分病例出现不同程度阳性表达,IDH-1及MGMT阴性,ATRX未缺失,EMA、PR、P53及CyclinD1均为阴性表达,Ki-67约1%~2%。CyclinD1在PA中的表达未见明显增加[13];PA缺乏IDH1突变、TP53突变和1p/19q共缺失,而常常出现BRAF基因激活[14]。60%~94%的PA出现BRAF和KIAA1549特异性融合,多发生于中线及幕下PA,小脑更高[15];FAM131B-BRAF基因的融合也有报道[16]。约2%~9%的PA患者可发生BRAFV600E基因突变,导致BRAF基因的持续激活[17]。少数PA患者也存在KRAS基因点突变,突变频率约为3%~5%[18]。
2.5 鉴别诊断 (1)弥漫型星形细胞瘤:影像学上表现为无边界的浸润性生长,MRI T1WI可为高信号或低信号,T2WI为高信号,增强扫描可见肿瘤强化,可囊性变;组织学上表现为一致性星形细胞肿瘤细胞,细胞密度稍大,血管增生,无Rosenthal纤维及嗜酸性小体,无双极性细胞区及多极细胞区,IDH1常突变。(2)少突胶质细胞瘤:脊髓内少突胶质细胞瘤极其罕见,影像学上很难与其他胶质瘤鉴别,微钙化是重要的表现,类似颅内少突胶质细胞瘤[19];组织学表现为肿瘤细胞较密集排列,细胞形态一致,核居中,胞浆透亮,似煎蛋样,分支状毛细血管,常伴有散在小灶钙化;分子遗传学表现为IDH1突变,1p/19q联合缺失。(3)室管膜瘤:影像学表现无特异性;组织学表现为血管周假菊形团,室管膜腔隙为特点,肿瘤细胞放射状排列于血管周围形成菊形团样,血管周围形成无细胞区;免疫组化为S-100及GFAP阳性表达,EMA为核旁点状阳性表达。(4)血管母细胞瘤:组织学表现毛细血管网支架内见间质细胞,间质细胞可呈巢状分布,胞浆透明样,可见脂质空泡;免疫组化为CD31、CD34、D2-40阳性表达,GFAP阴性表达。
2.6 治疗与预后 脊髓PA主要采用外科手术全切除肿瘤治疗,囊性变的肿瘤,需带囊壁一起切除,达到肿瘤全切的目的。脊髓PA患者术后是否需要行放疗尚存争议,但大多学者认为,PA为低级别星形细胞瘤无需放疗[20],即使复发,可再次行手术治疗。PA生长缓慢,患者术后10年的存活率>80%[21],术后神经功能的恢复为治疗关键。本组患者术后随访观察6~36个月,患者均健在且未发现肿瘤复发,症状均有不同程度的减轻,其中4例患者的症状明显好转,1例症状缓解并无加重。术前McCormick分级为Ⅰ级和Ⅱ级的患者术后症状恢复较快,预后较好;术前分级为Ⅳ级、病变累及C1-T7段的患者术后症状有所减轻,神经功能逐步恢复,但时间较长。
综上所述,脊髓PA少见,属于低级别胶质瘤(WHO Ⅰ级),临床表现为非特异性的肢体疼痛、麻木、无力;影像学检查无特征性表现,可伴有脊髓空洞形成;术前常被诊断为室管膜瘤或血管母细胞瘤,确诊需依靠病理学检查。PA的组织形态学特点结合免疫组化及分子学检测有助于诊断;其分子学检测表现为缺乏IDH1突变、TP53突变和1p/19q共缺失,并常出现BRAF基因突变。PA的治疗方法以手术全切除肿瘤为主,术后不需要行放疗,患者的神经功能经长时间的康复治疗可得到较好恢复,预后良好。
