应对国六排放的柴油机尾气NOx催化转化剂的试制研究
2021-02-26韦夏夏谭宗勇陈梦寅刘来君唐富顺孙强刘光文
韦夏夏,谭宗勇,陈梦寅,刘来君,唐富顺,孙强,刘光文
(1.桂林理工大学化学与生物工程学院材料科学与工程学院,广西 桂林 541004;2.广西辉煌朗洁环保科技有限公司,广西 北海 536000)
根据生态环境部发布的《中国机动车环境管理年报(2018)》,汽车保有量中柴油车占9.4%,其排放的NOx接近汽车排放总量的70%,PM超过90%,特别是占汽车保有量7.8%的柴油货车排放了57.3%的氮氧化物和77.8%的颗粒物,是机动车污染防治的重中之重。近几年我国柴油车的保有量呈现快速上升趋势,环境压力巨大。为此,我国相继推出了GB 18352.6—2016《轻型汽车污染物排放限值及测量方法(中国第六阶段)》(2020-07-01实施)、GB 17691—2018《重型柴油车污染物排放限值及测量方法(中国第六阶段)》(2019-07-01实施),对柴油车尾气的NOx排放相对于国五标准收严了80%,耐久性寿命要求不低于16万km,这对催化剂和净化器提出了极高的要求和挑战[1-5]。根据玉柴机器开发国六柴油机的瞬态工况测试循环对比WHTC/ETC试验情况来看,NOx排放工况点分布向低速工况偏移,国六柴油机的后处理装置更偏重于考核低排温工况,特别是低排温工况下NOx低温净化性能。
国六排放标准对装有钒基SCR(选择性催化还原)催化剂的车辆作出了规定,要求在正常寿命期内不得向大气中泄漏含钒化合物,并要求SCR 的入口温度低于550 ℃,但鉴于V2O5-WO3/TiO2催化剂的De-NOx性能较为稳定、SCR反应选择性高(N2O生成量较低)和抗水抗硫性能强[6-8],因而V2O5-WO3/TiO2催化剂仍然是柴油机尾气NOx排放控制满足国六排放标准的后处理技术之一。然而,钒基SCR催化剂的低温窗口特性窄[9-10],无法满足国六排放的要求,需对V2O5-WO3/TiO2催化剂进行生产技术的改进。本研究拟对V2O5-WO3/TiO2催化剂的制备技术进行研究,并改善其低温窗口特性,为国六排放柴油机NOx转化商用催化剂和净化器技术开发提供参考。
1 实验部分
1.1 V2O5-WO3/TiO2催化剂制备
锐钛矿型TiO2载体按中国发明专利(专利号:ZL 201310144882.4)制备:取一定量的湿偏钛酸,于100 ℃空气干燥12 h,650 ℃空气焙烧12 h,制得锐钛矿型TiO2载体,比表面积约为43.5 m2/g。搅拌条件下,称取一定量的草酸(AP),溶于去离子水中,加入适量的偏钒酸铵(AP)得绿色偏钒酸铵溶液。适量的偏钨酸铵(催化剂级)直接加去离子水溶解制备偏钨酸铵溶液。分别用分浸法(先W后V和先V后W)和共浸法制备V2O5-WO3/TiO2催化剂。将上述TiO2粉末载体浸入偏钨酸铵水溶液,置80 ℃水浴蒸干,100 ℃空气干燥12 h,500 ℃空气焙烧3 h,制得WO3/TiO2催化剂。再将WO3/TiO2催化剂浸入偏钒酸铵溶液,置80 ℃水浴蒸干,100 ℃空气干燥12 h,500 ℃空气焙烧3 h,制得先W后V的V2O5-WO3/TiO2催化剂。先V后W时,过程相反。共浸法时,TiO2载体浸入偏钨酸铵和偏钒酸铵混合溶液,条件同上。
1.2 V2O5-WO3/TiO2催化剂调变
将制备的锐钛矿型TiO2粉末加入到一定体积不同浓度的水合硝酸铈或水合氯化锡的水溶液中,80 ℃水浴蒸干,100 ℃干燥12 h,500 ℃空气气氛下焙烧4 h,得到改性MOx/TiO2氧化物。其中,所得SnO2/TiO2氧化物以80~100 ℃热水浸泡2 h后过滤,如此反复洗涤至滤液检测不出Cl离子后100 ℃干燥12 h。将改性所得MOx/TiO2氧化物以共浸渍法负载V2O5和WO3组分,条件同1.1节。
1.3 商用蜂窝催化剂制备
湿偏钛酸加入适量无机黏结剂、有机酸和水,球磨制成浆,并以真空涂覆的方法将浆料均匀附着于93 孔/cm2的陶瓷蜂窝孔内壁,催化剂干基负载量控制在不低于120 g/L。涂覆后于100 ℃空气干燥24 h,650 ℃空气焙烧12 h,制得锐钛矿型TiO2负载型陶瓷蜂窝催化剂载体。然后按1.2节的步骤负载助剂组分、V2O5和WO3组分,其中浸渍过程中蜂窝转动。
1.4 催化剂活性评价
6.2~9.3孔/cm2的粉末催化剂样品预先在500 ℃空气流(20 mL/min)处理30 min。SCR反应条件:0.1 MPa,原料气中NO为0.075%,n(NH3)∶n(NO)=1.05,O2体积分数为3.0%,其余为N2,混合气流量450 mL/min。反应前后NOx浓度用FGA10在线烟气分析仪检测。
蜂窝催化剂样品的SCR活性评价在排放后处理台架上进行,测量稳态工况下国六催化器研发样件不同温度时的NOx净化效率,测试方法按重型柴油车污染物排放限值及测量方法(中国第六阶段)(GB 17691—2018)进行。试验设备有YC6JA240-50发动机、ABB气体排放分析仪、奕科测功机、博世尿素喷射控制系统。
1.5 催化剂物性及结构表征
X射线衍射分析(XRD):所用仪器为Philips X’pert pro X射线衍射仪,测定条件为Cu 靶、Ni滤波片、管压40 kV、管流40 mA、扫描角度10°~80°。
BET比表面积测试:用Micrometrics ASAP2020比表面积测定仪测定样品的比表面积;N2吸附,-196 ℃。样品预先在300 ℃和真空度小于0.4 Pa的条件下进行脱气处理。
程序升温还原(H2-TPR):取0.1 g催化剂样品置于U型石英管中,用 (H2+Ar)混合气 (含H2体积分数5%) 流经催化剂样品,流速为20 mL/min。升温速率10 ℃/min。热导池响应耗氢信号,得到H2-TPR谱图。用CuO作外标对TPR谱图中的耗氢峰面积进行标定,以定量地计算催化剂的耗氢量。
NH3吸附-原位漫反射红外光谱(NH3-DRIFTS):采用赛默飞(Thermo Fisher)IS10傅里叶原位红外光谱仪,波速范围1 000~4 000 cm-1,扫描次数为16次,分辨率为4 cm-1,原位池窗片为KBr。取25 mg催化剂粉末,在N2气流(30 mL/min)中300 ℃恒温处理0.5 h,降至50 ℃后通入NH3+N2混合气(n(NH3)∶n(N2)=1% )(30 mL/min)至吸附饱和,切换N2气流(30 mL/min)吹扫1 h,以10 ℃/min速率升温至100 ℃,在不同温度下记录样品的红外光谱,得NH3-DRIFTS谱。
2 结果与讨论
2.1 V2O5-WO3/TiO2催化剂制备方式对NOx转化的影响
V2O5在锐钛矿TiO2表面分散容量的试验测定值约为1.14 mmolV/100 m2TiO2,与锐钛矿TiO2优先暴露晶面(001)的八面体空位密度(1.16 mmolV/100 m2TiO2)接近,认为分散态的钒离子可能键合在TiO2表面的八面体空位[11]。WO3在TiO2表面单层分散容量为0.75 mmolW/100 m2TiO2(即4.5 W/nm2)[12],按照“嵌入模型”,锐钛矿TiO2上优先暴露的晶面上,当嵌入W6+离子后与W6+相伴的O2-在表面形成密置单层[13-14]。图1示出300 ℃时各种催化剂下NO转化率随反应时间的变化。由图1可以看到,纯TiO2和WO3/TiO2催化剂样品的NO转化率很低(约3.0%),而V2O5/TiO2和V2O5-WO3/TiO2催化剂的NO转化率较高,约为22.0%和42.0%,且V2O5-WO3/TiO2催化剂的NO转化率明显高于V2O5/TiO2催化剂。这说明V2O5-WO3/TiO2催化剂VOx组分为SCR反应的主要催化活性组分,而V2O5-WO3/TiO2催化剂中WOx组分为助剂组分。随反应时间增加,V2O5-WO3/TiO2催化剂的NO转化率变化不大,表明该催化剂具有良好的催化稳定性。
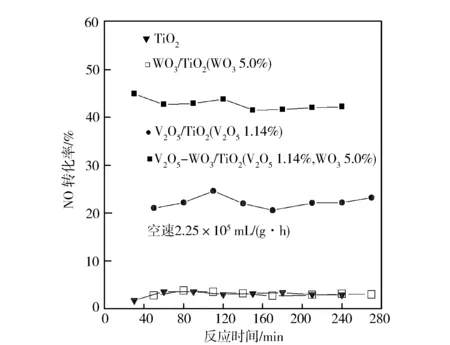
图1 催化剂的NO转化率(300 ℃)随反应时间的变化
图2示出先W后V分浸法制备的WO3负载量接近其分散容量(约0.69 mmolW/100 m2TiO2)时不同V2O5负载量(mmolV/100 m2TiO2)的V2O5-WO3/TiO2催化剂XRD谱图。从图2可见,V2O5负载量低于0.75 mmolV/100 m2TiO2,未出现晶相V2O5衍射峰,也未出现晶相WO3衍射峰(图2中b-e)。V2O5负载量超过0.75 mmolV/100 m2TiO2,可以观察到晶相V2O5衍射峰,且其强度随V2O5负载量增加而增加,但仍未观察到明显的晶相WO3衍射峰(图2中f-j),这说明后负载的V离子未从TiO2载体表面空位置换出已占据的W离子,V离子可能是在WO3/TiO2催化剂表面呈密置单层的WO3表面进行分散。
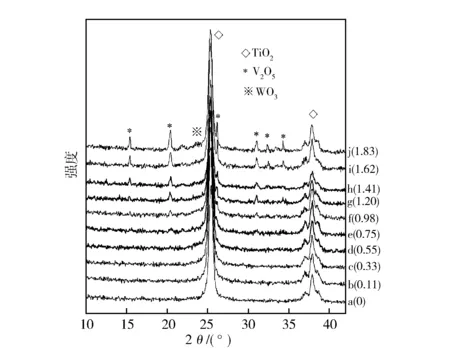
图2 不同V2O5负载量V2O5-WO3/TiO2催化剂(WO3 0.69 mmolW/100 m2TiO2) XRD谱图
图3示出先V后W分浸法制备的V2O5负载量略超过其分散容量(1.30 mmolV/100 m2TiO2)时不同WO3负载量(mmolW/100 m2TiO2)的V2O5-WO3/TiO2催化剂XRD谱图。从图3可见,WO3负载量低于0.58 mmolW/100 m2TiO2未出现晶相WO3衍射峰或晶相V2O5衍射峰(图3中b-e)。WO3负载量超过0.75 mmolW/100 m2TiO2时,可以观察到晶相WO3衍射峰,且其强度随WO3负载量增加而增加,但仍未观察到明显的晶相V2O5衍射峰(图3中f-j)。这说明后负载的W离子也未能从TiO2载体表面空位置换出先期已占据 的V离子,W离子可能是在V2O5/TiO2催化剂表面分散。
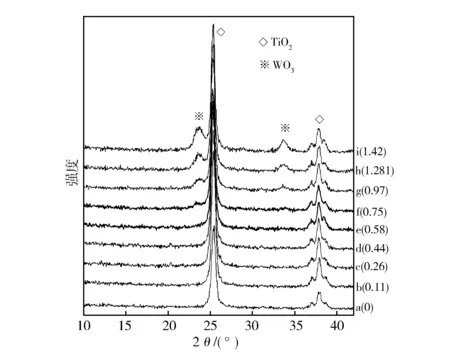
图3 不同WO3负载量V2O5-WO3/TiO2催化剂(V2O5 1.30 mmolV/100 m2TiO2) XRD谱图
图4示出共浸法制备的V2O5和WO3负载量同比例增加的V2O5-WO3/TiO2催化剂XRD谱图。从图4可见,V2O5和WO3负载量分别为1.30 mmolV/100 m2TiO2和0.69 mmolW/100 m2TiO2的V2O5-WO3/TiO2催化剂其XRD谱图上未出现晶相V2O5和晶相WO3的衍射峰。当V2O5和WO3负载量分别同比例增加1.5倍和2.0倍时,XRD谱图上可观察到相应的晶相V2O5和晶相WO3的衍射峰,且晶相V2O5和晶相WO3的衍射峰强度随V2O5和WO3负载量的增加相应地增加。这说明超量部分的VOx和WOx物种可能在催化剂表面聚积形成晶相的V2O5和晶相WO3。

图4 共浸法制备不同负载量(mmol/100 m2TiO2)V2O5-WO3/TiO2催化剂XRD谱
表1列出不同方法制备的V2O5-WO3/TiO2催化剂SCR活性。从表1可见,在催化剂组成相同的情况下,催化剂SCR活性依次为共浸法、分浸法(先W后V) 、分浸法(先V后W)。如前所述,后负载的W离子不能从TiO2载体表面空位置换出先期已占据的V离子,后负载的V离子也不能从TiO2载体表面空位置换出先期已占据的W离子,故分浸法制备V2O5-WO3/TiO2催化剂WOx物种和VOx物种分散均匀性较差、相互作用较弱,故催化剂SCR活性较低。并且,先V后W分浸法制备的V2O5-WO3/TiO2催化剂WOx物种对部分表面VOx物种的覆盖,使得催化剂SCR活性更低。而共浸法制备的V2O5-WO3/TiO2催化剂,WOx物种和VOx物种在载体表面分散均匀,二者之间存在较强的相互作用,从而具有较高的SCR活性。
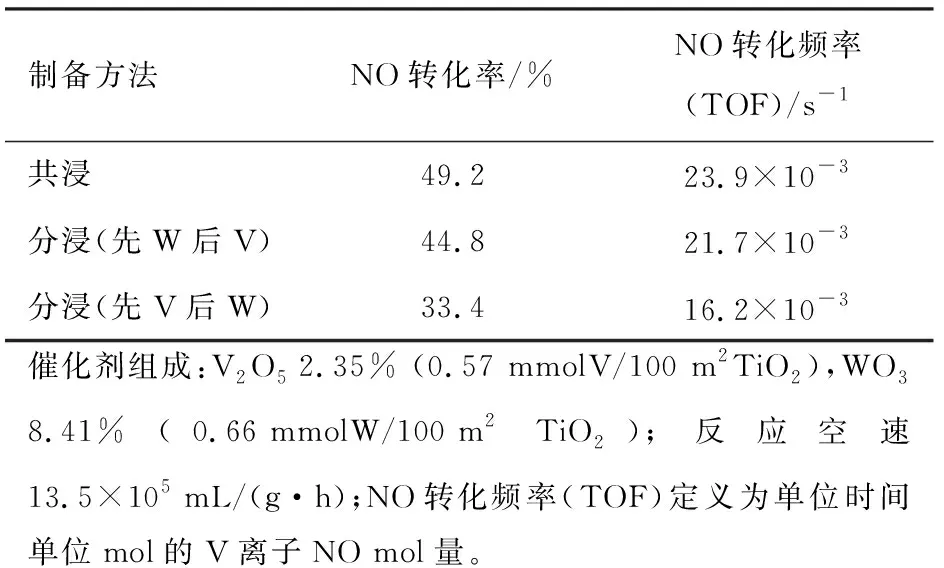
表1 不同方法制备的V2O5-WO3/TiO2催化剂SCR活性(300 ℃)
2.2 V2O5-WO3/TiO2催化剂的低温SCR性能优化和工况试验
图5示出分别以10%CeO2和10%SnO2调变1%V2O5-3%WO3/TiO2催化剂后的SCR活性。由图5可以看到,反应空速3.75×104mL/(g·h)下,以共浸渍法制备的1%V2O5-3%WO3/TiO2催化剂其T50约为275 ℃,尽管其350 ℃以上的高温SCR性较好,但其低温活性窗口较窄。当1%V2O5-5%WO3/TiO2催化剂以10%CeO2和10%SnO2分别调变后,催化剂的T50分别向低温迁移到约190 ℃和210 ℃,同时,T90亦分别同步向低温迁移,这说明V2O5-WO3/TiO2催化剂中掺杂CeO2和SnO2组分,可以显著改善低温SCR性能。但也可以看到,掺入量相同时,CeO2组分对V2O5-WO3/TiO2催化剂的低温活性窗口改善要比SnO2组分的更佳。

图5 CeO2和SnO2调变的V2O5-WO3/TiO2催化剂SCR活性
图6示出 CeO2和SnO2组分调变的V2O5-WO3/TiO2催化剂的X射线衍射(XRD)、程序升温还原(H2-TPR)和NH3吸附-原位漫反射红外光谱(NH3-DRIFTS)分析结果。从图6a可知,以10%CeO2调变V2O5-WO3/TiO2催化剂后,28.5°可见微弱的CeO2衍射峰,说明催化剂上有少量CeO2组分聚集生成了晶相CeO2,另一方面也表明大部分CeO2组分可高分散于催化剂表面。同时,以10%SnO2调变V2O5-WO3/TiO2催化剂后,催化剂上没有观察到晶相SnO2衍射峰,表明SnO2组分高分散于催化剂表面。另可见,掺杂CeO2和SnO2组分调变均使V2O5-WO3/TiO2催化剂的25.3°的锐钛矿相TiO2(A)衍射峰变弱,其中SnO2组分对TiO2锐钛矿晶相结构影响更为明显。这说明CeO2和SnO2组分均与TiO2发生了较强的相互作用。更为显著的是,SnO2组分与TiO2的强相互作用,导致了催化剂在27.4°出现了明显的金红石相TiO2(R),表明SnO2组分降低了TiO2晶型转变温度,促进锐钛矿型TiO2向金红石型TiO2转变。无论是CeO2组分或SnO2组分调变的V2O5-WO3/TiO2催化剂上均没有观察到V2O5和WO3的衍射峰,这说明V2O5和WO3仍高度分散于催化剂上,同时也进一步表明以共浸渍法负载的V2O5和WO3组分其分散稳定性较好。
从图6b可见,以10%CeO2调变V2O5-WO3/TiO2催化剂后,尽管Ce和V离子的还原峰均略向高温偏移(555 ℃),但还原峰面积明显增大,还原程度仍然是增加的,其原因应为555 ℃的还原峰应包含了Ce和V离子的还原叠加。与此不同的是, V2O5-WO3/TiO2催化剂以10%SnO2调变后,V离子的还原峰向高温偏移至580 ℃,且峰面积下降,然而,该催化剂在320 ℃的还原峰面积增大。这表明Sn与V的作用可使部分V离子更容易还原,离子的还原能力有所增强。H2-TPR的结果表明了CeO2和SnO2组分对V2O5-WO3/TiO2催化剂的可还原性影响不同。尽管CeO2和SnO2组分都使V2O5-WO3/TiO2催化剂上V离子的还原温度向高温偏移,但CeO2组分还原可使催化剂的低温还原耗氢量增加,其还原性增加来源于CeO2组分可还原性,有可能CeO2组分也作为活性位点参与了SCR反应,而SnO2组分却促进了部分V离子低温还原能力的增强,进而促进催化剂的低温SCR性能。
进一步考察CeO2和SnO2组分调变的V2O5-WO3/TiO2催化剂表面酸性(见图6c),可以看到,V2O5-WO3/TiO2催化剂上呈现Lewis酸位(1 172 cm-1)和Brnsted酸位(1 450 cm-1)[15-16],掺杂CeO2和SnO2组分后均使V2O5-WO3/TiO2催化剂上的Brnsted酸位上NH3红外吸光值峰面积增大,表明掺杂CeO2和SnO2组分促进了V2O5-WO3/TiO2催化剂上Brnsted酸性增强。根据Topse[15]的Brnsted酸SCR反应机理,Brnsted酸有利于SCR反应的进行。因此,V2O5-WO3/TiO2催化剂上掺杂CeO2和SnO2组分可使催化剂表面的Brnsted酸位更多,促进了SCR反应的进行。

图6 CeO2和SnO2调变的V2O5-WO3/TiO2催化剂的XRD、H2-TPR和NH3-DRIFTS图谱
综上可见,利用共浸法制备V2O5-WO3/TiO2催化剂,更能促进WOx物种和VOx物种在TiO2载体表面分散均匀性和稳定性,从而具有较高的SCR活性。V2O5-WO3/TiO2催化剂上掺杂CeO2和SnO2组分后,可显著增加催化剂表面Brnsted酸位,有利于提高催化剂的低温SCR反应性能。CeO2组分的低温还原性和SnO2组分促进了部分V离子低温还原能力的增强也是提高催化剂低温SCR性能的原因之一。由于SnO2组分与TiO2的强相互作用促进锐钛矿型TiO2向金红石型TiO2转变,低温可还原性相对弱,这可能是导致SnO2组分掺杂对V2O5-WO3/TiO2催化剂的低温活性窗口改善要比CeO2组分相对差的原因。
综合上述试验结果,以共浸渍法制备了CeO2和SnO2等优化调变的V2O5-WO3/TiO2蜂窝催化剂样品(催化剂涂层中V2O5组分质量分数约为0.5%),并在排放后处理台架上进行了稳态工况下NOx净化效率试验,台架测试发动机为玉柴YC6JA240-50柴油机,测试结果见表2。依据本研究结果所制备的蜂窝催化剂样品,其不同工况下的NOx转化效率均达到96%以上,SCR催化性能稳定,低温转化效率较高,应可以满足国六排放的要求。当然,该催化剂的耐久性需要进一步的试验考察。
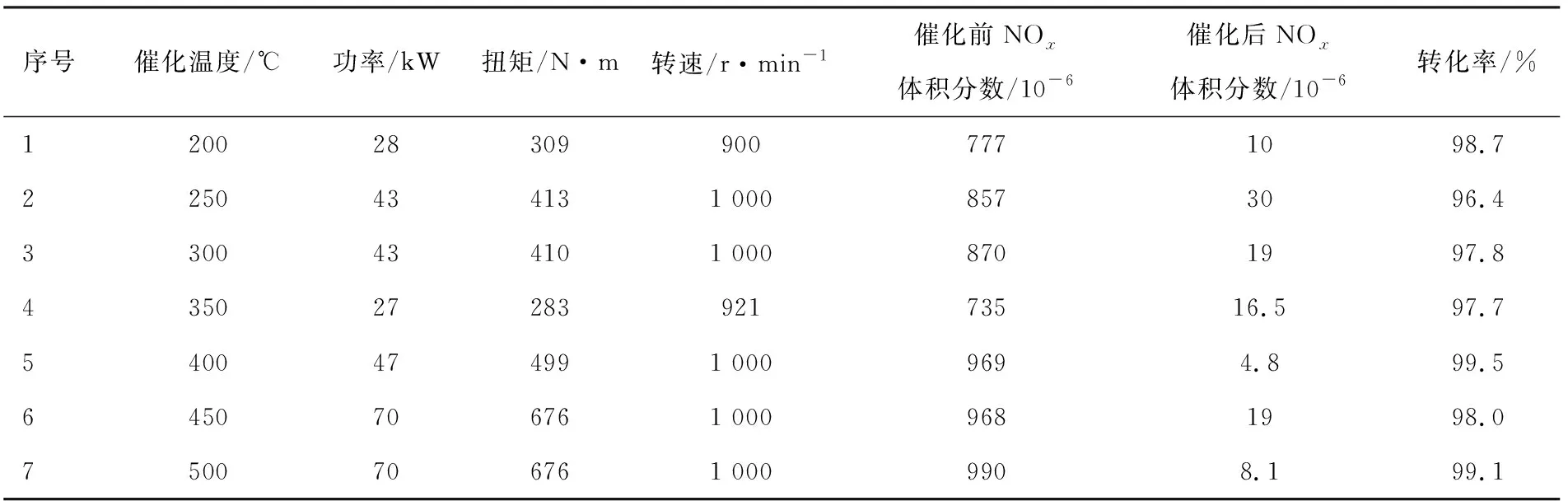
表2 稳态工况下国六催化器不同温度时的NOx净化效率
3 结束语
活性组分的负载方式对V2O5-WO3/TiO2催化剂SCR性能影响较大,在催化剂组成相同的情况下,催化剂SCR活性依次为共浸法、分浸法( 先W后V)、分浸法( 先V后W)。共浸法制备的V2O5-WO3/TiO2催化剂,WOx物种和VOx物种在载体表面分散均匀,相互作用较强,具有较高的SCR活性。CeO2和SnO2组分调变V2O5-WO3/TiO2催化剂可以显著改善其低温SCR性能。稳态工况试验表明,共浸渍法制备的CeO2、SnO2调变型V2O5-WO3/TiO2蜂窝催化剂样品具有稳定优异的低温SCR催化性能。
