气溶胶稳定硫同位素研究进展
2021-02-17牛振川黄智浦
马 皓 ,王 森 ,牛振川, ,冯 雪, ,黄智浦
1.
西北大学 城市与环境学院,西安 710127
2.西北大学 陕西省地表过程与环境承载力重点实验室,西安 710127
3.陕西西安城市生态系统定位观测研究站,西安 710127
4.中国科学院地球环境研究所 黄土与第四纪地质国家重点实验室,西安 710061
5.中国科学院第四纪科学与全球变化卓越创新中心,西安 710061
6.陕西省加速器质谱技术及应用重点实验室,西安加速器质谱中心,西安 710061
7.陕西关中平原区域生态环境变化与综合治理国家野外科学观测研究站,西安 710061
8.西安地球环境创新研究院,西安 710061
硫酸盐是大气气溶胶的重要组成部分,是雾霾和大气酸化的重要因素,硫酸盐细颗粒更是云凝结核的重要来源,经干湿沉降导致环境酸化,引起一系列的生态环境问题(Adams et al,1999;王代长等,2003;Bao and Reheis,2003;郑珂等,2019;Sun et al,2019)。还可通过直接和间接辐射强迫作用,影响全球气候变化(Charlson et al,1992;Kiehl and Briegleb,1993;孙家仁和刘煜,2008)。此外,硫酸盐气溶胶还会引起呼吸系统疾病,对人类健康产生不利影响(Ostro et al,2010;王瑞霞,2018)。
大气环境中硫酸盐气溶胶可分为人为和自然活动排放的一次硫酸盐气溶胶和二次硫酸盐气溶胶。后者是大气硫酸盐气溶胶的重要成分,是由化石燃料燃烧、火山活动和生物活动排放的含硫气体(SO2、H2S和二甲基硫化物等)的最终氧化产物(Lelieveld et al,2001;Li et al,2013;Li et al,2020)。目前硫酸盐气溶胶源解析的方法主要有模型法(Itahashi et al,2012;Itahashi et al,2019),标记示踪法(张延君等,2015;Itahashi et al,2017)等,但这些方法易受环境因素影响,且只能大致判断污染源的来源,对污染源的定量表达较差。相比之下,同位素溯源技术具有测量精度高、对源解析准确和测量误差小等优势,采用稳定硫同位素组成来有效示踪大气中硫的来源和循环过程已取得一系列进展(郭照冰等,2010;郭照冰等,2014;Han et al,2016;Han et al,2017;Chung et al,2019)。局部区域的稳定硫同位素组成往往具有均一性和特定性,而不同硫源的稳定硫同位素组成存在差异,通过这种差异可以分析不同介质中硫的来源、迁移和转化过程(Norman et al,2006;Ghahremaninezhad et al,2016;Chen et al,2017)。通过对硫酸盐气溶胶稳定硫同位素的研究,可以了解大气硫同位素的形成机制,更好地定量各硫源的贡献率,揭示不同硫源对大气气溶胶的影响(Norman et al,2004a;Guo et al,2016;Chung et al,2019)。因此,本文系统地综述了稳定硫同位素的分析方法,总结其在不同地区的分布特征,探究气溶胶硫酸盐稳定硫同位素的示踪研究及其形成过程中的同位素分馏特征,并展望未来稳定硫同位素技术在气溶胶溯源方面的应用,为控制气溶胶硫酸盐污染提供科学依据。
1 稳定硫同位素的简介与主要分析方法
1.1 稳定硫同位素的表示方法
硫在环境中有四种稳定的同位素:32S、33S、34S、36S,其相对丰度分别为95%、0.74%、4.2%和0.016%(Berglund and Wieser,2011)。最早以美国代阿布洛大峡谷铁陨石中的陨硫铁(canyon diablo troilite,CDT)为硫同位素国际标准,因CDT样品缺乏,故目前常采用国际原子能机构(IAEA)提出的V-CDT(Vienna-canyon diablo troilite)为标准,取IAEA-S-1(一种人工合成的Ag2S)相对于V-CDT的值δ34S为−0.3‰,硫同位素组成用δ值来表示,即:

式中:xS代表稳定硫同位素种类(33S、34S、36S)(Stichler et al,1995)。
硫同位素分馏分为动力学非平衡分馏、热力学平衡分馏和非质量相关分馏,前两者的分馏过程均与质量相关,即δ33S、δ34S和δ36S之间存在如下定量关系(Hulston and Thode,1965):

而非质量分馏导致δ33S和δ36S 偏离了上述质量分馏定律,通常用∆33S和∆36S表示,其计算公式如下(Hulston and Thode,1965;Farquhar and Wing,2003):
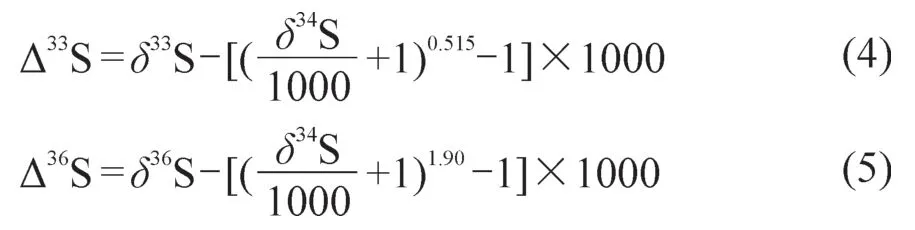
当∆33S和∆36S不等于0时,就认为存在硫同位素的非质量分馏效应。
1.2 气溶胶稳定硫同位素的分析方法
气溶胶稳定硫同位素的分析方法主要分为两类(表1):一类是整体样品分析,气溶胶样品经前处理成为纯硫化物,再通过质谱仪测量硫同位素;另一类是微区原位分析,即对单颗粒气溶胶样品进行分析。

表1 稳定硫同位素分析方法Tab. 1 Detection methods for stable sulfur isotopes
传统的气溶胶整体样品分析方法是先将硫化物氧化为SO2或SF6气体后再进行质谱分析(Coleman and Moore,1978;Rees,1978)。SO2进样时,样品先在高温纯氧环境中氧化燃烧为SO2后,再进入质谱仪分析。Pichlmayer and Blochberger(1988)提出用连续同位素质谱法测定硫同位素,将元素分析仪(EA)与稳定同位素比质谱仪(IRMS)联用,建立在线连续流分析方法。该方法实现了SO2的自动燃烧和色谱纯化,满足快速、高效测定硫同位素的要求(Giesemann et al,1994;Grassineau et al,2001;Studley et al,2002)。
通常,SO2进样的EA-IRMS法所需硫化物样品量为0.2 — 1.5 mg,较大的需样量使得珍贵样品难以满足其分析要求(Giesemann et al,1994;韩娟等,2018)。减少其样品需求量成为目前改进EA-IRMS测试方法的一个重要方向。武晓珮等(2020)在常规硫同位素分析装置上增加了一个六通阀和一个自动加热冷阱,并增加氦载气流速,样品利用率由0.3%提高至3%,需硫量降至30 —150 μg,测量值与真值差异在0.4‰以内。此外,其测量过程中不能很好地约束氧同位素组成的变化,δ34S值较标准值高1‰ — 3‰,难以精准测量33S和36S(Fry et al,2002)。
F只有一种同位素,样品以SF6形式引入时不受同位素干扰,能精准测量33S和36S。含硫物质依次转化为H2S和Ag2S,最后经氟化系统以SF6形式进入质谱分析。大气气溶胶研究中使用Thode solution(氢氟酸、次磷酸和盐酸混合液)将含硫物质转化为H2S,该反应液可长期保存但反应时间 较 长(Arnold et al,2014;Lin and Thiemens,2020)。SF6法样品量大于0.1 μmol时,Δ33S、δ34S和Δ36S的分析精度通常为0.01‰、0.1‰和0.1‰(Yang et al,2016;Jovovic et al,2020)。
除EA-IRMS外,近几年多接收电感耦合等离子体质谱仪(multi-collector inductively coupled plasma mass spectrometer,MC-ICPMS)应用于气溶胶硫同位素的分析显著增加。MC-ICPMS方法所需的样品硫低至100 pmol时,Δ33S与δ34S分析精度为0.2‰;当样品量硫为数百pmol时,Δ33S与δ34S的分析精度可达0.1‰和0.07‰(Das et al,2012;Kümmel et al,2020)。常与激光消融、液相色谱和气相色谱等联用,包括液体进样和固体进样两种方式(Lin et al,2014;Martínez-Sierra et al,2015)。目前对于气溶胶样品的分析常使用溶液进样的方式,可同时精准测量经HNO3溶液消解和离子树脂纯化的气溶胶样品中32S、33S和34S(Craddock et al,2008;Paris et al,2013)。此外,可通过调试将基体效应、质量歧视和谱学干扰等影响因素降低(Clough et al,2006;Craddock et al,2008;Zakon et al,2014;Pribil et al,2015;Liu et al,2016;卞霄鹏,2017)。Clough et al(2006)最先建立了利用MC-ICPMS测量硫同位素的方法,并采用硅标准溶液来校正硫的质量歧视效应。Das et al(2012)采用了阴离子交换树脂对样品进行净化,具有程序空白低、化学要求少和样品处理时间短等优势,建立了MC-ICPMS在低硫样品δ34S测量中的方法,可用于冰川、雪、雨水和气溶胶样品的研究。
二次离子质谱(SIMS)能测定微区范围内硫同位素的比值,是微区地球化学研究的重要工具。该方法摒弃了复杂耗时的湿法化学消解过程,对固体样品直接进样分析,采用铯离子轰击样品产生二次离子S−,通过多个法拉第杯在多接收模式下测试量同位素(Chen et al,2015;范宏瑞等,2018)。测量过程中仪器分馏和矿物化学成分引起的基体效应等因素使获得的同位素比值不能直接代表样品硫同位素比值,这需要研发新的标准样品来解决,因此标样的研发是SIMS发展的一个重要方向(李秋立等,2013;范宏瑞等,2018)。目前已建立了黄铁矿、黄铜矿和火山玻璃等样品硫同位素分析技术,研制了含硫矿物的标准物质以应对测量过程的基体效应(Li et al,2019;Shimizu et al,2019)。Kozdon et al(2010)采用Cs+离子源(光斑10 μm)对几微米尺度内黄铜矿、方铅矿和黄铁矿进行了原位硫同位素分析,其δ34S分析精度分别为0.3‰、0.3‰和0.2‰;将冲击能量由20 keV降至13 keV时,闪锌矿的测量精度由1.7‰提高到0.6‰。现今阶段,SIMS测量33S和36S还有较大误差,通常只能用于有明显非质量分馏的样品,如地学研究中的太古代样品。在分析精度未能得到有效改善的情况下,SIMS在现代大气气溶胶的研究潜力还相对有限。
新型二次离子质谱仪的发展提供了高精度和高空间分辨率下原位硫同位素原位测量的机会,如适合高精度分析的Cameca IMS—1270 / 1280和适合高空间分辨率测量的Cameca NanoSIMS等(李秋立等,2013)。气溶胶样品稳定硫同位素原位分析常采用纳米二次离子质谱技术(NanoSIMS),以具有高空间分辨率的Cameca NanoSIMS 50L型二次离子探针质谱仪为主。NanoSIMS离子探针技术可以对离子直径小于500 nm的单个含硫颗粒的多种硫同位素进行分析,实现S同位素纳米空间的测定(Winterholler et al,2006;Winterholler et al,2008a;Hauri et al,2016)。该方法在5 μm的空间分辨率下,δ34S的精度为0.3‰(杨蔚等,2015)。单颗粒气溶胶的同位素分析可以识别硫酸盐化学形态,以此为基础细化气溶胶硫酸盐的贡献。Winterholler et al(2008b)通过对比气溶胶整体样品和单颗粒样品硫同位素组成,分析了不同硫酸盐颗粒的来源及其对整体样品的贡献,认为海盐粒子中约含7.5%硫,而与Ca相关的硫酸盐来源于道路粉尘和建筑排放的一次CaSO4以及云内矿物粉尘表面形成的二次CaSO4。根据硫酸盐的化学成分或形态特征来进一步分析人为源硫和生物源硫在矿物粉尘表面反应和云内生成等化学反应,NanoSIMS法可应用于自然气溶胶和人为气溶胶的复杂混合物分析(Winterholler et al,2006;Winterholler et al,2008b)。
2 硫酸盐气溶胶稳定硫同位素组成特征
2.1 硫酸盐气溶胶δ34S的空间分布特征
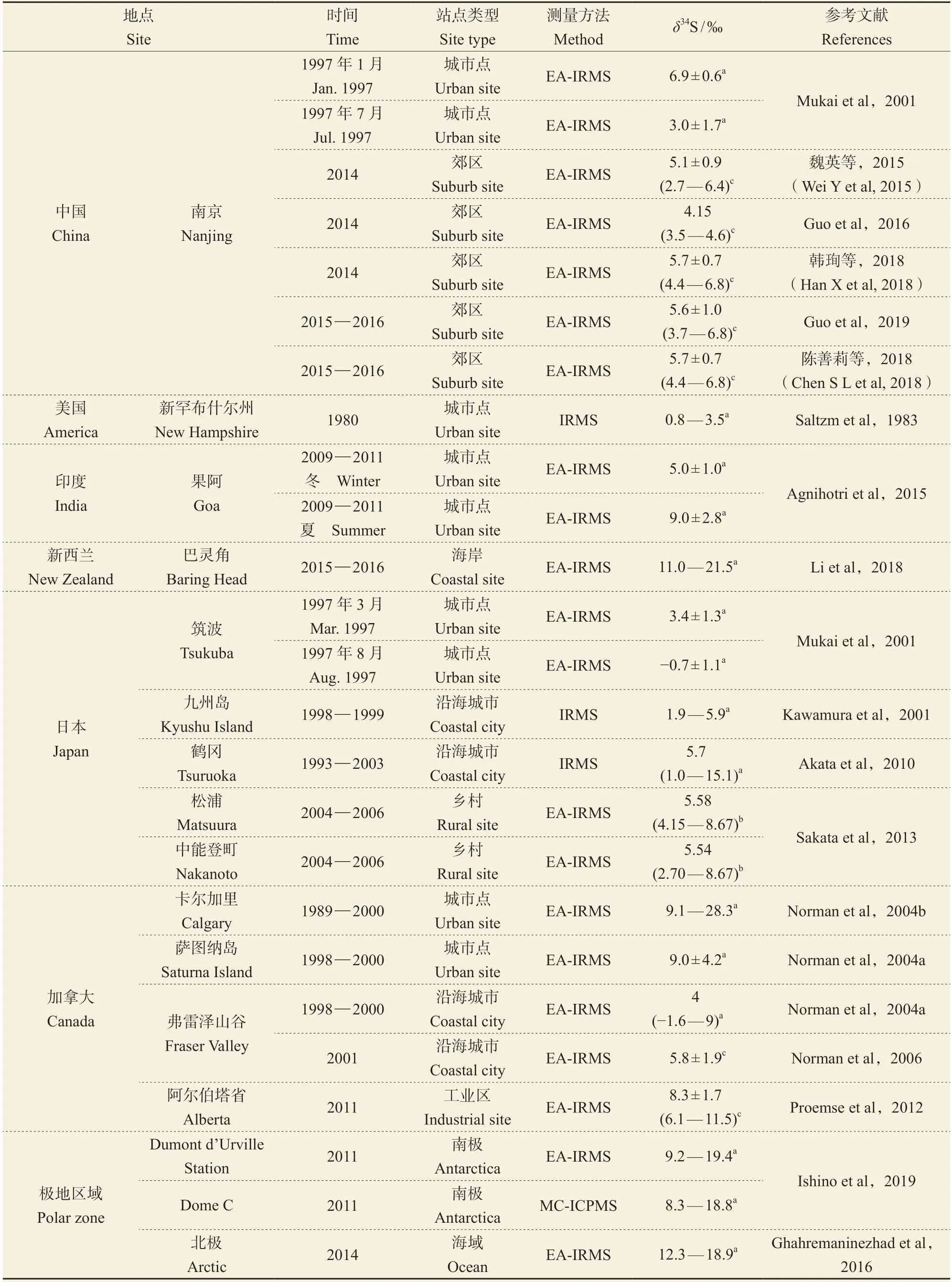
从空间尺度分析,全球硫酸盐气溶胶δ34S值在−1.6‰ — 28.3‰,气溶胶硫酸盐的δ34S在南北极地区和部分沿海区域值较高,在内陆区域值较低(表2),硫酸盐气溶胶来源δ34S组成的不同是造成其空间差异的主要原因。南北极区域(Ghahremaninezhad et al,2016;Ishino et al,2019)和新西兰Baring Head(Li et al,2018)地区受海洋生物硫和海盐作用明显,其气溶胶δ34S值偏高,集中在10‰ — 20‰。而另外一些沿海城市,如日本Tsuruoka、Matsuura、Nakanoto和加拿大Fraser Valley、Alberta等地,海洋来源与化石燃料燃烧来源均不容忽视。当气团主要来自海洋时,研究区域内海盐颗粒占比重,δ34S会出现较高值,接近15‰(Akata et al,2010);而在海洋气团较弱时,δ34S则更接近当地化石燃料硫同位素(Kawamura et al,2001;Mukai et al,2001;Norman et al,2004b;Norman et al,2006;Proemse et al,2012;Sakata et al,2013)。相 比沿海区域,内陆区域硫同位素变化主要受化石燃料燃烧影响。美国New Hampshire(Saltzm et al,1983)、印度Goa(Agnihotri et al,2015)以及中国大部分城市气溶胶δ34S组成均与当地化石燃料燃烧有关。中国北方城市如北京、天津、宜昌、哈尔滨等气溶胶δ34S值主要集中在3‰ — 8‰,南方城市如成都、南京、贵阳等地区δ34S值主要集中在1‰ — 6‰,北方城市气溶胶δ34S值略高于南方城市,这与中国北方煤炭δ34S(3.89‰)高于南方(−1.24‰)相一致(Hong et al,1993;Mukai et al,2001;Guo et al,2010;张苗云等,2011;郭照冰等,2012;Yang et al,2015;陶月乐,2018;Yang et al,2019b)。

表2 全球硫酸盐气溶胶中的稳定硫同位素水平Tab. 2 Stable sulfur isotopic levels in sulfate aerosols
此外,研究发现气溶胶粒径差异对δ34S值影响较小。当粒径大于0.15 μm时,δ34S值变化不大;粒径为0.15 — 3 μm的气溶胶颗粒与粒径大于7.2 μm的气溶胶颗粒的δ34S值差异仅为0.9‰(Ghahremaninezhad et al,2016)。对比2012 —
2015年北京市TSP和PM2.5,发现其δ34S值均在6‰上下波动(Guo et al,2010;Guo et al,2014;Han et al,2016;Han et al,2017;Wei et al,2018)。
(续表2 Continued Tab. 2)
此外,远距离传输会影响当地气溶胶硫同位素的空间分布(Karnieli et al,2009)。δ34S研究表明:中国燃煤排放硫经远距离迁移,对日本区域气溶胶有着较大贡献(Ohizumi et al,2016)。日本Matsuura和Nakanoto地区冬季非海盐硫酸盐气溶胶值为6.3‰ ± 1.1‰,这与中国北方冬季煤炭燃烧δ34S(6.7‰ ± 0.8‰)相近,这表明中国北方冬季煤炭燃烧可能是该地区的潜在硫源(Sakata et al,2013)。Nriagu et al(1991)发现1983—1985年北极区域气溶胶δ34S值与挪威、瑞典等北欧国家气溶胶δ34S值相似,结合大气后向轨迹认为北极区域大气硫大部分来源于欧洲。此外,同一地区还可能受到两个甚至多个方向硫传输的影响。Wei et al(2018)利用轨迹聚类和Flexpart法分析了北京硫酸盐气溶胶受远距离迁移的影响,认为δ34S值较轻(4.6‰ ± 0.8‰)的硫酸盐来自南方和东方,而值较重(6.7‰ ± 1.6‰)的硫酸盐来自北方和西北。因此,稳定硫同位素在揭示气溶胶硫酸盐的远距离传输来源中有重要作用。
2.2 硫酸盐气溶胶δ34S的时间变化特征
同时代硫同位素具有相同的δ34S值,同地区气溶胶稳定硫同位素值在数年期间不会发生重大变化。北京地区2005 — 2015年气溶胶δ34S均值在6‰左右(Guo et al,2010;Guo et al,2014;Han et al,2016;Han et al,2017;Wei et al,2018),南京地区2014 — 2015年气溶胶硫同位素均值在5.1‰ — 5.7‰(Guo et al,2016;Chen et al,2017;陈善莉等,2018;韩珣等,2018),但人为活动会影响硫同位素组成。Akata et al(2010)分析了1993—2003年日本Tsuruoka大气硫酸盐气溶胶δ34S值的变化,发现自2000年以后气溶胶δ34S的值有明显变化,从0‰ — 15‰降至0‰ — 5‰,这与湿式石灰石法烟气脱硫工艺引入当地焦炭生产工业和热电厂有关。该工艺流程中硫同位素发生分馏,燃烧副产物富较重硫同位素,释放的SO2气体富集轻硫同位素,使得气溶胶的δ34S组成偏低(Derda et al,2007)。
此外,气溶胶δ34S存在季节性变化。日本沿海城市非海盐气溶胶硫酸盐(Kawamura et al,2001;Akata et al,2010;Sakata et al,2013)和中国北京(Han et al,2016;Han et al,2017)、成都(Yang et al,2019b)等地气溶胶硫酸盐δ34S值都呈现出冬季高于夏季的变化趋势。造成气溶胶δ34S季节性变化的原因是:(1)冬季煤炭燃烧释放富重硫同位素的SO2,而夏季陆地生物源排放硫化物的同位素较轻(刘广深等,1996;李亲凯等,2016)。(2)SO2氧化途径的季节性变化。冬季以异相氧化为主,硫酸盐富重硫同位素,夏季以均相氧化为主,硫酸盐富轻硫同位素(Saltzman et al,1983;Tanaka et al,1994)。同时,不同氧化途径中硫同位素质量分馏效应也会受温度影响(Harris et al,2013a),实验表明温度每升高1℃,硫酸盐硫同位素下降0.08‰(Caron et al,1986)。与上述季节变化特征相反的是,印度西部Goa地区夏季气溶胶δ34S值(9‰)明显高于冬季(5‰),这是因为该地区夏季气溶胶大部分来源于邻近阿拉伯海海洋生物排放富重硫同位素的含硫气体(Agnihotri et al,2015)。此外,因夜间比日间温度低,影响了SO2的氧化方式,使得夜间气溶胶δ34S值较高于日间(Yang et al,2015;陶月乐,2018;Yang et al,2019b)。
3 硫酸盐气溶胶形成过程中稳定硫同位素的分馏特征
3.1 气溶胶34S的质量分馏
以往硫同位素溯源是建立在从排放源到气溶胶硫酸盐形成过程中硫同位素不发生分馏的基础上,而SO2的氧化过程(均相氧化和异相氧化)伴随着富重硫同位素的平衡分馏和富轻同位素的动力学分馏,SO2和硫酸盐气溶胶硫同位素值存在差异(图1)(郭照冰等,2010)。SO2氧化生成的硫酸盐占多数时,硫酸盐δ34S受同位素分馏效应变化明显,不能直观指示出污染源(郭照冰等,2010)。为了细化气溶胶硫酸盐的来源,需要对SO2氧化过程中硫同位素的分馏进行评估,分析两种氧化机制对二次硫酸盐的贡献率。常以分馏系数α表示SO2和其生成物硫酸盐之间的同位素分馏,从而判断不同氧化途径的相对重要性(Saltzman et al,1983;Chen et al,2017),即:

图1 硫酸盐气溶胶物化过程中δ34S的变化及其不同来源δ34S范围Fig. 1 Variation in δ34S during the physicochemical process of sulfate aerosols and the δ34S range of different sources

SO2在以羟基自由基为氧化剂的均相氧化过程中仅存在动力学分馏,其氧化反应必然导致硫酸盐富集轻同位素,氧化过程中34S的过渡态临界能的正差导致分馏系数小于1(Tanaka et al,1994)。但这种变化会被过渡态更紧密的振动流形抵消,34S分馏系数可能大于1(Leung et al,2001)。最近研究表明,SO2以为氧化剂时,分馏系数为1.0089 ± 0.0007 − [(4 ± 5)×10−5]T(℃)(Harris et al,2012)。
在云和液滴中,以H2O2、O3、NO2和过渡金属离子(transition metal ion,TMI)为氧化剂的异相氧化过程中,硫同位素存在两种分馏,但总体上动力学分馏效应要远小于平衡分馏(Saltzman et al,1983)。平衡分馏氧化反应的产物则会富集重硫同位素,δ34S值升高约16.5‰,分馏系数大于1(Eriksen,1972)。H2O2与O3为氧化剂时,34S的液相氧化分馏系数为1.0167 ± 0.0019 − [(8.7 ±3.5)×10−5]T(℃),研究表明云内酸碱度会使分馏系数升高,生成物硫酸盐相比SO2会富集34S(15.1‰ — 19.9‰)(Harris et al,2012;Harris et al,2013b)。NO2氧化SO2时,硫同位素分馏系数表现出明显的温度效应,当温度小于33℃时,生成物富集重硫同位素;当温度大于33℃时,重硫同位素被消耗;温度为8 — 33℃时,分馏系数大于1(Yang et al,2018)。TMI催化氧化时,生成物硫酸盐富轻硫同位素34S(−9‰ ± 3.1‰),TMI氧化过程温度效应为(0.237‰ ± 0.004‰) ∙ ℃−1(Harris et al,2013a;Harris et al,2013b)。研究表明19℃时TMI的催化氧化的分馏效应αFe为0.9894 ± 0.0043(Harris et al,2012)。
一些学者在假定硫酸盐气溶胶全部来源于SO2氧化和氧化过程只发生硫同位素质量分馏的前提下,基于如下公式评估了SO2非均相氧化和均相氧化对硫酸盐气溶胶的相对贡献(Seal,2006;张苗云等,2011;Guo et al,2016)。
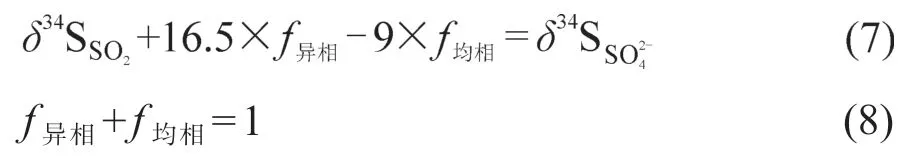
Chen et al(2017)发现南京冬季SO2氧化成硫酸过程中,α值为1.0014 — 1.0075,异相氧化的贡献率为40.7% — 64.8%;而在南京夏季青奥会期间,α值范围为1.0024 — 1.008,均相氧化贡献率为51.3%,略高于异相氧化的贡献率47.3%(Guo et al,2016)。分馏系数变化可归因于硫同位素分馏的温度效应。实验研究表明:作为氧化剂时α的温度效应((0.004‰ ± 0.015‰) ∙ ℃−1)明显低于H2O2和TMI作为氧化剂时的温度效应((0.085‰ ± 0.004‰) ∙ ℃−1,(0.237‰ ± 0.004‰) ∙ ℃−1)(Harris et al,2013a)。Winterholler et al(2008b)分析了二次硫酸盐的硫同位素比值,结合欧洲历年数据发现异相氧化对硫酸盐形成的贡献从20世纪90年代初的50%增加到2005年的60% — 70%。研究者认为主要是与SO2的O3氧化过程有关,一方面因为大气O3的浓度增加,以O3为氧化剂的SO2氧化贡献率增加,另一方面则是因为O3氧化速率与pH有关,欧洲大气pH从早期的4.4变为4.9,O3氧化速率增加。
气溶胶的δ34S值一方面与污染源有关,污染源的种类改变或贡献率变化会引起气溶胶δ34S改变,但这种变化可能更多地影响初次硫酸盐的δ34S。另一方面与SO2的氧化过程有关,主要影响二次硫酸盐颗粒δ34S,尤其是在二次硫酸盐污染严重的城市。最近一项利用气溶胶硫同位素评估北京市2015年污染源贡献的研究中发现,污染天和非污染天的污染源贡献量没有明显变化,但是其δ34S有差异,这种差异是由于SO2氧化途径的变化引起的(Fan et al,2020)。因此,气溶胶硫同位素δ34S的变化是显示了排放源的变化还是氧化途径的变化,可能需要具体分析初次硫酸盐和二次硫酸盐的贡献率及其δ34S,以此来判断哪种方式影响了气溶胶δ34S的变化。
3.2 气溶胶硫同位素的非质量分馏
气溶胶硫同位素还存在非质量分馏效应。认识气溶胶硫同位素的非质量分馏效应,有助于探讨燃烧反应过程中硫同位素分馏机理,分析城市气溶胶硫酸盐的形成机制,了解大气硫的循环过程(Romero and Thiemens,2003;马红梅等,2010;Thiemens and Lin,2019)。早期学者认为城市气溶胶硫同位素非质量分馏与平流层SO2的光化学氧化有关。在小于310 nm波段紫外线的影响下,平流层SO2光解氧化生成硫酸盐气溶胶过程中发生硫同位素非质量分馏现象,随后平流层大气传输至对流层中,影响对流层气溶胶硫同位素(Farquhar et al,2000;Farquhar et al,2001;Savarino et al,2003)。研究发现中国太湖流域硫酸盐气溶胶Δ33S变化范围为0.081‰ — 0.386‰,Δ36S范围在−1.24‰ — −0.14‰,Δ33S和Δ36S不存在显著相关(郭照冰等,2012);北京地区PM2.5中硫酸盐Δ33S在春、夏和秋三个季节为0.300‰ — 0.480‰(Wei et al,2018)。上述研究中发现的气溶胶硫同位素非质量分馏(即Δ33S和Δ36S不等于0)可归因于平流层SO2光化学氧化中产生非质量分馏后向对流层的传输;实验室模拟研究表明SO2在紫外线氧化过程中存在明显的正Δ33S和负Δ36S关系(Masterson et al,2011)。
最近的一些研究表明,燃烧过程和矿物颗粒表面的非均相反应也可导致硫同位素非质量分馏。北京市2015 — 2016年冬季气溶胶Δ33S<0‰,研究者认为一方面是因为冬季煤炭的不完全燃烧过程发生Δ33S<0‰的非质量分馏,这种硫同位素特征会转移至大气中;另一方面则与北京市冬季气象条件有关,北京冬季的逆温现象阻止了平流层的输入(Lee et al,2002;Han et al,2017)。郭照冰等(2014)研究了北京气溶胶中多硫同位素的非质量分馏效应,结合对位有效能(CAPE)分析发现其形成机制除与平流层SO2光化学氧化反应有关外,还可能与含硫化合物的燃烧有关(Sun et al,2019)。Lin et al(2018a)采用放射性同位素35S及其他四种稳定硫同位素综合分析气溶胶硫同位素异常的原因,认为Δ36S<0与平流层的光氧化和煤炭本身无关,Δ36S异常可能与化石燃料或生物质燃烧过程中形成的初级硫酸盐有关。Lin et al(2018b)基于早期的研究和同位素质量平衡认为生物质燃烧会出现Δ33S异常(−0.67‰ ± 0.20‰),2015 — 2016年北京市污染期间也观测到相似的气溶胶Δ33S(−0.664‰)异常(Han et al,2017)。与燃烧相关的过程会导致气溶胶Δ33S和Δ36S异常,这可能是燃烧过程中基于量子对称效应的硫重组反应(如SH+H→S + H2、SH + SH→S + H2S、S+SH→S2+ H等)可能导致非质量分馏的出现,这种反应与质量无关(Yan et al,2005;Babikov,2017;Lin et al 2018b;Thiemens and Lin,2019)。此外,SO2在矿物粉尘颗粒表面的非均相氧化反应也是硫同位素非质量分馏的一个可能因素。Yang et al(2018)和Yang et al(2019a)研究认为SO2通常的氧化(TMI +H2O2和NO2)过程中的硫同位素分馏并不能解释城市气溶胶Δ33S的异常,其异常跟平流层向对流层传输也无关,可能跟额外氧化方式有关。近期Genot et al(2020)的研究指出,Δ33S的异常可能与矿物颗粒表面的非均相反应有关。综上所述,硫同位素非质量分馏的机理仍有许多不确定性,基于实验室和现场观测来继续研究大气气溶胶多硫稳定同位素非质量分馏的机理仍是未来重要的研究方向。
4 硫酸盐气溶胶稳定硫同位素示踪研究
利用硫同位素自然丰度变化,对比各种可能存在硫源的δ34S就可以确定研究区域硫酸盐气溶胶的来源。当潜在硫源不多于两种的时候,利用δ34S可以定量分析硫源的贡献率(Moncaster et al,2000);当硫源大于两种时,可以定性分析硫源,缩小来源范围(Shanleya et al,2008)。
4.1 人为成因硫
煤炭燃烧和交通排放是城市区域大气硫污染的主要来源(Wadleigh et al,1996)。在世界范围内,煤的硫同位素范围在−30‰ — 30‰,而中国煤炭同位素在−10‰ — 10‰(Hong et al,1993)。中国南方城市煤炭δ34S相对较低,为− 1.24‰,北方煤炭硫同位素平均值为3.89‰。郭照冰等(2014)发现北京大气气溶胶δ34S值集中在5‰ — 6‰,与北京地区煤中硫同位素组成(4.75‰)接近。表明煤炭燃烧是中国城市硫酸盐气溶胶的重要来源(Hong et al,1993;Motoyama et al,2011)。Sakata et al(2013)研究发现日本西海岸气溶胶受冬季东北风影响,可能主要来自于中国北方燃煤。
除煤炭燃烧外,交通排放也是城市气溶胶的重要硫源。研究发现2014 — 2015年南京市气溶胶δ34S值为2.7‰ — 6.8‰,其值在当地煤炭燃烧(4.6‰ — 6.6‰)和汽车尾气(4.6‰ — 9.7‰)的δ34S值范围之间,表明南京市硫酸盐气溶胶主要来源于当地燃煤和汽车尾气排放(韩珣等,2018)。Yang et al(2015)在利用气溶胶质量比确定固定污染源和移动污染源的基础上,对比气溶胶δ34S发现冬季宜昌市颗粒物的主要来源是固定源燃煤(δ34S,3.68‰)和移动源汽车尾气(δ34S,5‰)的混合排放。而国外一些城市,如新西兰Baring Head、加拿大Calgary等地区汽车尾气排放是二次硫酸盐的主要来源。研究发现新西兰地区商业航运的排放物δ34S(3‰ ± 3‰)与气溶胶硫同位素组成(3‰)几乎一致(Li et al,2018)。加拿大Calgary地区汽车尾气中SO2和细气溶胶硫酸盐的同位素组成分别为9.3‰和10‰,汽车尾气是其气溶胶硫酸盐主要来源(Norman et al,2004b)。总体而言,中国大部分城市气溶胶硫酸盐主要来源煤炭燃烧,而交通排放可能是国外许多发达城市的重要来源。
4.2 自然成因硫
4.2.1 生物成因
生物硫主要来自陆地生物和海洋生物,其δ34S值相差较大。陆地生物硫δ34S值范围在− 10‰ — 5‰(Nriagu et al,1987;刘广深等,1996;Mast et al,2001;Norman et al,2004a)。夏季生物新陈代谢活动较冬季旺盛,生物成因硫作用明显,温哥华地区生物成因硫酸盐贡献率可高达44%(Norman et al,2004a)。生物硫的释放影响颗粒物的硫同位素组成,富轻同位素导致了夏季大气硫同位素组成的降低,这也是造成气溶胶δ34S季节性变化的重要原因(Nriagu et al,1987;Mast et al,2001;Guo et al,2016)。Han et al(2016)等利用δ34S值探索北京气溶胶硫的来源,发现生物硫是夏季气溶胶中硫的主要来源。
海洋生物硫酸盐会影响沿海区域大气硫同位素组成,对其二次硫酸盐贡献率可高达79%(Arimoto et al,1996;Li et al,2018)。海洋上空由浮游植物释放出的二甲基硫化物(DMS)经氧化形成的硫酸盐气溶胶δ34S值为15‰ — 19‰(Calhoun et al,1991;Wagenbach,1996)。Norman et al(2004a)等发现加拿大Saturna岛气溶胶δ34S值与海洋DMS的δ34S值相近,并计算了DMS对加拿大硫酸盐气溶胶的贡献,发现海洋生物产生的硫酸盐在Saturna岛中占非海盐硫酸盐的30%,是其主要来源。在北极地区,夏季海洋生物对直径小于0.49 μm的细颗粒硫酸盐气溶胶的贡献率 高 达63%(Ghahremaninezhad et al,2016)。 Li et al(2018)利用硫同位素分析确定了南大洋二次硫酸盐的来源:海洋生物DMS生成的硫酸盐占二次硫酸盐的90%,这表明海洋生物活动是控制海洋大气中硫酸盐气溶胶含量的重要因素。
4.2.2 海盐成因
海盐硫酸盐δ34S值高于海洋生物DMS,约为20‰ — 21‰(Thode et al,1961;Rees et al,1978)。海洋上空由海洋喷雾产生的海盐硫酸盐通过气团迁移,会对部分沿海地区大气硫酸盐δ34S产生一定影响(Kawamura et al,2001)。研究发现南北极海域,沿海城市如日本Matsuura和Nakanoto等地区气溶胶受海盐气溶胶影响,导致δ34S值偏高0.2‰ — 1.3‰(Sakata et al,2013)。基于δ34S质量平衡方程可评估出非海盐硫酸盐(non-sea salt sulfate)值δ34Snss:

4.2.3 火山成因
火山喷发的SO2仍是全球硫的重要来源之一,与其他来源相比较,火山气溶胶可在平流层停留长达一年(Andres and Kasgnoc,1998;Elias and Sutton,2012)。火山喷发早期的硫酸盐气溶胶富轻硫同位素,而后期富重硫同位素(de Moor et al,2010)。大型火山喷发时期含硫气体传输至平流层区域,SO2经光化学氧化反应发生非质量分馏,Δ33S可以用来辨别火山喷发期间平流层大气SO2的氧化反应和迁移过程(Baroni et al,2007)。Bindeman et al(2007)分析火山灰中硫酸盐同位素发现大型火山喷发时,火山灰硫酸盐Δ33S与δ34S总体呈负相关,Δ36S与δ34S呈正相关,而小型火山喷发时则无相关性,这可能跟大型火山喷发SO2在平流层通过气相氧化生成硫酸盐,而小型火山喷发SO2在对流层气溶胶表面发生氧化反应有关。Martin(2018)用硫氧多同位素系统研究火山硫酸盐,发现不同途径的SO2氧化有可能发生在岩浆、对流层或平流层中。
5 结论与展望
本文对气溶胶硫酸盐硫同位素的分析方法、分布特征、分馏机理和其在硫酸盐来源示踪中的应用进行了总结和归纳,有如下主要结论:
(1)EA-IRMS适用于多硫同位素的高精度分析,MC-ICPMS适用于低硫样品分析,NanoSIMS适用于不需要化学前处理的单颗粒气溶胶微观分析。
(2)气溶胶稳定硫同位素空间分布组成受污染源和远距离迁移作用影响,δ34S空间分布可简单归纳为:远洋区域>沿海区域>内陆区域。δ34S的季节变化普遍呈冬高夏低,与冬夏季节不同的排放源和SO2的氧化方式有关。
(3)气溶胶稳定硫同位素存在质量分馏和非质量分馏,质量分馏发生与SO2的氧化过程有关,影响气溶胶硫同位素组成;后者归因于SO2光化学氧化、燃烧过程和矿物颗粒表面的非均相反应。
(4)以煤炭燃烧和汽车尾气为主的人为成因硫是许多内陆城市硫酸盐气溶胶的主要来源,夏季生物成因硫的贡献也不容忽视。而海盐硫酸盐和火山硫酸盐通过大气传输作用对其周边区域气溶胶组成影响较大。
目前气溶胶硫同位素的非质量分馏和δ34S变化所代表的意义等研究上仍有许多不同观点,建议今后的研究应该加强以下几方面工作:
(1)加强气溶胶硫同位素非质量分馏研究。一方面加强平流层向对流层输入硫的观测,另一方面深入分析气溶胶硫同位素非质量分馏的原因,除SO2的光化学反应、燃烧和矿物表面氧化外,是否还有其他可能的原因还值得未来进一步深入的研究。
(2)丰富硫同位素示踪模型,同位素示踪技术应考虑从来源至气溶胶颗粒的大气化学过程中硫同位素分馏变化。大气δ34S的变化是显示了排放源变化还是SO2氧化途径的变化,是现今学界的重要争论,也这是未来的重要研究方向之一。此外,贝叶斯模型已经较多应用于大气同位素研究中,可以将贝叶斯模型和硫酸盐硫氧双同位素结合起来精确量化不同污染源对大气硫酸盐的贡献,为明确大气硫的环境效应和分馏机制提供科学依据。
