谈GMP文件体系及其管理
2021-01-11冼海燕
冼海燕
摘要:良好的文件是质量管理体系的重要组成部分,是符合药品生产质量管理规范(GMP)要求的关键。GMP文件体系中的各项文件应包括充分的指导细节,以便于执行者对标准和要求有着相同的理解;同时还应提供对各种过程的充分记录,以便证明既定的标准和要求是持续被遵守的。
关键词:GMP文件体系;文件管理
【中图分类号】 R-1 【文献标識码】 B 【文章编号】2107-2306(2021)17--01
GMP,是“药品生产质量管理规范”的Good Manufacturing Practice英文名称简称。GMP(2010年修订版)第一百五十条指出:“文件是质量保证系统的基本要素。必须有内容正确的书面质量标准、生产处方和工艺规程、操作规程以及记录等文件。”也就是说,按规定建立GMP文件体系,用通俗的行话讲,“写你做的,做你写的,记你做的”。主要目标是建立、控制、监测和记录直接或间接影响药品质量各方面的所有活动,以确保药品生产的各项活动持续遵守既定的标准和要求。那么,GMP文件体系包括哪些内容及其要求又有哪些?
1.GMP文件体系制定时的一般原则包括:
1.1所有可能影响药品特性、质量的生产、包装、存储、销售和实验室的操作,以及执行GMP的辅助性文件(如清洁记录、预防性维护报告等)都必须有书面文件,且由质量管理部门审核和批准。
1.2GMP文件必须符合GMP、中国药品管理法等法规、规范的要求。
1.3建立GMP文件体系前,必须明确与文件制定、批准和使用相关的角色和及其职责。文件审核过程必须确保文件准确、完整、符合法规要求,所下的结论是有科学依据的。
1.4起草、审核和批准GMP文件的人员必须对相应的设备和工艺有适当的技术理解。
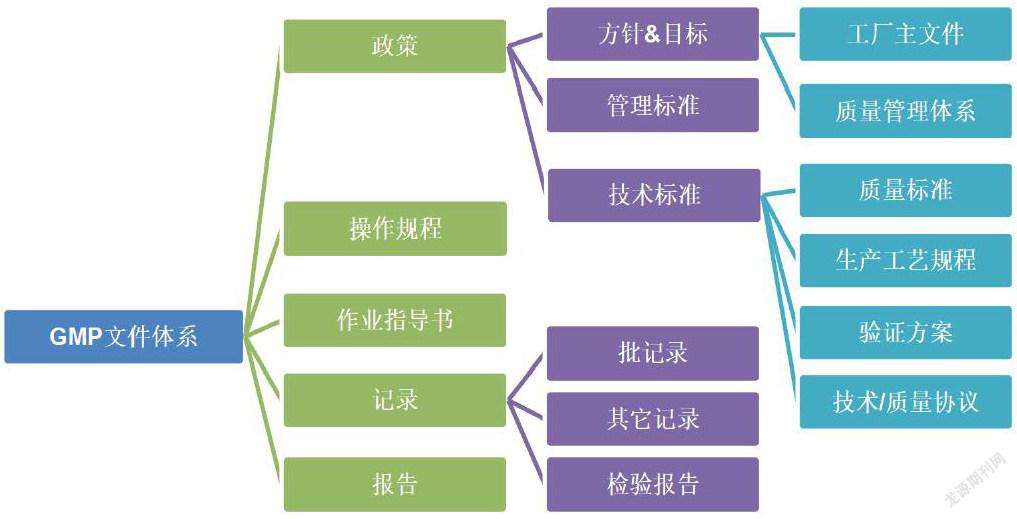
2.GMP文件体系的构成
2.1建议GMP文件体系有以下文件:
2.1.1工厂主文件:描述工厂与执行GMP相关的所有活动。
2.1.2质量管理体系,是以文件化的形式建立了实现有效质量管理所需的组织结构、职责和规程的正式体系。质量管理体系至少应建立确保产品质量和患者安全所需的文件,包括但不限于政策、程序或记录,例如:
·人员和培训
·文件管理
·生产和过程控制
·标签和包装控制
·来料验收
·纠正和预防行动
·变更管理
·质量审计
·过程质量监控
·储存和发运
·投诉处理
·年度报告
2.1.3指引类文件:包括技术标准、管理规程和操作规程,例如:
2.1.3.1质量标准:详细描述生产过程中使用或制得的产品或物料必须符合的要求。
2.1.3.2生产处方和工艺规程:提供所有要使用的起始原料、设备和计算机系统的详细信息,并说明所有工艺、包装、取样和检验操作说明。同时,应规定要采用的过程控制和过程检验技术以及可接受标准。
2.1.3.3操作规程&作业指导书,例如确认和验证、校准、设备的维护及清洁和消毒、培训、更衣、环境监测、虫害控制、偏差处理、药品召回、退货等。
2.1.3.4方案:给出执行和记录某些特定操作的说明,如验证方案。
2.1.3.5技术/质量协议:由委托方和受托方就GMP外包活动达成的协议。
2.1.4记录和报告
2.1.4.1记录:提供各项活动符合操作说明的证据,例如活动、事件、调查。记录还包括用于生成其它记录的原始数据。
2.1.4.2批记录:是记录每批产品的历史记录。
2.1.4.3检验报告书:提供产品或物料样品的检验结果及结论。
2.1.5报告:记录特定活动、项目或调查的进行情况,以及结果、结论和建议。包含不仅限于以下内容:
·多产品共用的可行性评估报告
·确认或验证报告
·持续稳定性考察报告
·现场质量审计报告
·定期的质量回顾分析报告
·召回报告
·GMP自检报告
·年度报告
2.2 GMP文件体系
3.GMP文件的控制
3.1应有文件定义文件类型,并且有文件管理规程。
3.2随着科学技术的发展和应用,许多文件如操作规程或作业指导书和记录等,可能以电子文件和纸质文件的形式共存。此类情况,应说明原文件、正式复制件、数据处理和记录的关系及控制措施。
3.3应制定控制措施对电子文件实施适当的控制,以确保记录在整个保存期内的完整性。
3.4 GMP变更控制规程应包含GMP文件的修订,该变更必须由经授权的人员进行。
3.5 GMP文件变更控制包括:
3.5.1控制所有文件的发布、修订、替换和撤销,以确保提供和使用正确的文件。
3.5.2修订历史的使用和维护。
3.5.3在使用新修订的文件之前,必须对该文件进行培训并记录。
3.6如使用电子数据处理系统、照相技术或其他可靠方式记录数据资料,应当有所用系统的操作规程;记录的准确性应当经过核对。
3.7使用电子数据处理系统的,只有经授权的人员方可输入或更改数据,更改和删除情况应当有记录;应当使用密码或其他方式来控制系统的登录;关键数据输入后,应当由他人独立进行复核。
4. 良好文件规范
4.1所有记录的手写内容必须清晰,并用不褪色的笔书写。
4.2 GMP文件中的签名必须清晰可辨,以便可追溯到执行该步骤的人员。在适当的情况下,电子签名可以代替手写签名。
4.3记录应当保持清洁,不得撕毁和任意涂改。
4.4应遵照数据可靠性原则。
4.5在执行每个操作和/或发生需要记录的事件时都应填写记录。必须根据相应的 GMP 文件的职责对记录的内容进行检查、确认、审核和/或批准。
4.6必须管理和控制记录原件,以确保将不当使用和伪造记录的风险降低到可接受的水平。
5.数据可靠性原则
在数据的整个生命周期中,记录必须是可追溯的、清晰的、及时的(在发生时记录)、原始的和准确的(ALCOA)。
5.1可追溯的(Attributable):应能确定执行记录任务的人。需记录谁执行了任务、谁对记录做了更正、删除、更改等。
5.2清晰的(Legible):所有的记录都必须是清晰易读的。
5.3及时的(Contemporaneous):应在行动、活动或决策发生时进行记录。
5.4原始的(Original):原始记录可以描述为第一次获取的信息,无论是纸质记录还是电子记录。
5.5准确的(Accurate):通过诸多因素的实施能确保结果和记录的准确性。
6.GMP文件的保存
6.1必须保存所有GMP文件。
6.2文件必须以安全、可靠的方式保存,并可快速检索。
6.3记录必须完整。
6.4记录必须仅供授权的用户查阅或访问。
6.5在要求的保存期内必须保证GMP文件或记录的数据的完好性和完整性。
6.6批记录的保存期限至少按照监管规范的规定执行;而其它类型的文件的保存期限,取决于文件所支持的业务活动,包括在上市许可有效期内,保留作为上市许可信息支持的原始数据(例如与验证或稳定性相关的数据)。
6.7文件的保存期限应在内部操作规程中规定。
【讨论】
良好的文件是质量管理体系的重要组成部分,是符合药品生产质量管理规范(GMP)要求的关键。GMP文件体系中的各项文件应包括充分的指导细节,以便于执行者对标准和要求有着相同的理解;同时还应提供对各種过程的充分记录,以便证明既定的标准和要求是持续被遵守的。
参考文献:
[1] 药品生产质量管理规范(2010年修订)(卫生部令第79号) 2011.1.17发布
[2] MHRA.GMP Data Integrity Definitions and Guidance for Industry.Jan,2015
[3] FDA.Data Integrity and Compliance with CGMP Guidance for Industry.Apr,2016
[4] Model of Organization, Management and Control, referred to in the Italian Legislative Decree 231/01 and subsequent amendments.
