镅锔分离研究进展
2020-12-30朱礼洋李晓敏杨素亮张生栋
朱礼洋,李晓敏,2,杨素亮,张生栋,*
1.中国原子能科学研究院 放射化学研究所,北京 102413; 2.兰州大学 核科学与技术学院,甘肃 兰州 730000
镅(Am)和锔(Cm)有着特殊的用途。241Am目前大部分应用于烟雾探测报警器;在物体厚度测量及密度测量等方面也有应用,与Be构成Am-Be中子源可用于油井测井[1]。242Cm和244Cm可用于放射性同位素电池[2]。Am、Cm材料获取、高放废液处理和超钚元素生产均涉及Am、Cm分离技术。乏燃料经PUREX流程处理后,产生的高放废液的放射性贡献主要来自90Sr和137Cs,经过200~300 a后基本衰变完全,其后热量来源主要来自Am[3]。Am和Cm留在高放废液中约需10 000 a放射性水平才能降到天然铀的水平,如果将Am和Cm分离,则只需几百年即可达到天然铀水平。因此将Am和Cm从高放废液中进一步分离,可减少最终固化废物的热源,有利于简化最终地质处置。在高放废液管理中Cm的长期放射性贡献几乎可以忽略[4],因此法国原子能机构(CEA)的路线倾向于仅分离出Am而将Cm留在高放废液中。Cm的大部分同位素放射性强、嬗变产率低,在ADS嬗变系统中,去除生热的Cm同位素可以降低嬗变燃料制备的难度,另外只含有Am的燃料对于嬗变反应堆的要求也更低。
生产超钚元素时也经常涉及到Am、Cm的化学分离与纯化。生产高纯度238Pu的一个途径是中子辐照241Am生成242Cm,然后将242Cm与Am进行分离,得到的242Cm放置一段时间后通过α衰变即可得到高纯度的238Pu[5]。在252Cf生产中,也涉及Am、Cm、Bk、Cf的分离问题[6]。因此,非常有必要进行Am和Cm的分离研究。
1 Am和Cm的基本性质
目前已知Am的同位素有19种,质量数为232~247,半衰期为55 s~7 370 a。已知Cm的同位素有15种,质量数为237~251,半衰期为2.3 h~156万年[7-8]。燃耗33 000 MW·d/t压水堆乏燃料冷却10 a后,每吨铀(MTU)含有约594 g Am(503 g241Am、0.66 g242Amm和90.6 g243Am)和18.9 g Cm(0.34 g243Cm、17.6 g244Cm和0.93 g245Cm)。对放射性贡献较大的Am、Cm同位素是241Am、243Am和244Cm,放射性会随时间下降,但是241Am会随241Pu的衰变而增加[1]。
锕系与镧系元素类似,也有类似的半径收缩效应[9],Seaborg等[10]总结了三价、四价镧系和锕系离子的离子半径与原子序数的关系。三价镧系离子的4f电子较靠近内层,受核电荷的影响较大,因此4f电子在外层区域出现的几率低,镧系离子和配体的轨道重叠很少。而锕系的5f电子具有一定的延展性,在配位时具有一定的共价性[11]。Am原子的电子结构是[Rn]5f77s2, Cm原子的电子结构是[Rn]5f76d17s2,对三价离子,Am(Ⅲ)是5f6,而Cm(Ⅲ)是高稳定性的5f7。
Am具有+3、+4、+5和+6四种氧化态,五价和六价Am的构型与U、Pu、Np锕酰离子类似[12]。据推测,采用脉冲辐解并辅以光谱检测,可以检测到短寿命物种。在含K2S2O8的8 mol/L KOH的Am溶液中,采用N2O饱和后冷冻到-60 ℃,可能会检测到更高价态的Am[13]。Am(Ⅲ)晶体离子半径约为1.00 Å(1 Å=0.1 nm),而Am(Ⅳ)的离子半径约为0.85 Å[14],相差较大。Cm 具有+3和+4两种氧化态。Cm(Ⅲ)的电子构型较为稳定,其还原和氧化电位均很高,Cm(Ⅲ)/Cm(Ⅱ)电对的电位约为-2.78 V,很难将Cm(Ⅲ)还原,而Cm(Ⅳ)/Cm(Ⅲ)电对的电位约为-3.1 V,需要非常强的氧化剂才能将Cm(Ⅲ)氧化[1]。
2 Am的氧化分离方法
通过氧化剂或电化学方法可将Am(Ⅲ)氧化到Am(Ⅳ)、Am(Ⅴ)或者Am(Ⅵ),继而容易实现Am和Cm的分离。Mincher等总结了Am(Ⅳ)、Am(Ⅴ)和Am(Ⅵ)的制备条件[15],并综述了美国有关高价态Am的制备、表征和分离研究[16]。总的来说,Am(Ⅲ)的氧化剂种类有限,且生成的高价态Am也易被还原。Am(Ⅳ)较难处理,防止其歧化的措施会增加体系复杂性,因此鲜有基于此价态的分离方案。Am(Ⅵ)的还原速率太快,会产生Am(Ⅴ)和Am(Ⅲ)的混合价态,基于Am(Ⅵ)的分离流程也存在不少问题。Am(Ⅴ)的络合能力差,目前基于Am(Ⅴ)的分离研究还较少。
采用NaBiO3固体可以将Am(Ⅲ)氧化到Am(Ⅵ)或Am(Ⅴ),这取决于不同的温度。大量研究表明,将Am(Ⅲ)氧化到Am(Ⅵ)后,可进行流程规模的萃取。通常分配比(D(Am))可达到3~4左右,但是偶尔也会比预期的低,小于2,这可能是因为试剂中杂质的还原性造成的。采用戊基膦酸二戊酯(DAAP)作萃取剂,在6~7 mol/L HNO3时D(Am)最高,Am(Ⅵ)的回收率约为65%~85%。在热实验中发现,萃取到有机相的Am(Ⅵ)在反萃过程中无法被充分还原,可能需要增加还原剂H2O2的浓度[16]。NaBiO3氧化法的缺点是动力学较慢,萃取前需批式操作氧化1~2 h,并且萃取前需要将多余的固体NaBiO3过滤。将Am(Ⅲ)氧化到Am(Ⅵ)后也可与硝酸铀酰共结晶,在晶体相中Am(Ⅵ)的稳定性会显著提高,13 d内未见明显变化,而水相中的Am(Ⅵ)经过10 d会有一半自动还原到Am(Ⅲ)[17]。美国内华达大学的Richards等[18]发现,NaBiO3固体除了具有氧化Am(Ⅲ)的作用外,在低酸时还可以选择性吸附Cm(Ⅲ)。NaBiO3固体具有钛铁矿结构,Cm(Ⅲ)与其表面上带负电的O有很强的作用,而氧化到高价态的Am与O作用较弱。直接采用NaBiO3固体作为吸附剂、Celite 535作为填充剂制成萃取色层,在0.1 mol/L HNO3中Am和Cm的分离因子可达90,对Am、Cm的回收率可达到97%,纯度达到90%。
在1~3 mol/L HNO3中采用银离子/臭氧可定量氧化Am(Ⅲ)到Am(Ⅵ),且Ag(Ⅰ)和Ag(Ⅱ)均不会被DAAP萃取,Ag可以循环使用。然而在使用1 mol/L DAAP萃取时,接触时间15 s时Am的分配比只有0.33,这与同样条件下萃取Am(Ⅲ)的结果一样,说明即使有过量Ag(Ⅱ)存在,在与有机相接触时,Am(Ⅵ)和Ag(Ⅱ)均不稳定[16]。与Ag/臭氧体系类似的高碘酸铜体系中,氧化剂三价铜可迅速定量将Am(Ⅲ)氧化到Am(Ⅵ),在c(HNO3)≤3 mol/L时Am(Ⅲ)的氧化率可以达到98%~99%。短暂的萃取,例如5 s的萃取D(Am)略低于2,说明有Am(Ⅵ)被萃取了,但是随萃取时间延长D(Am)反而下降,这是因为没有支持氧化剂存在,在与有机相接触时Am(Ⅵ)又被还原了[16]。
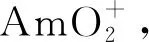
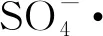
可见将Am(Ⅲ)氧化到Am(Ⅳ),存在易歧化的问题。氧化到Am(Ⅵ)是可行的,也是研究较多的方案,但在后续萃取时,价态往往难以保持。将Am氧化到Am(Ⅴ)进而实施Am(Ⅴ)与Cm(Ⅲ)的分离,往往可获得预期结果。
3 配位分离方法
配位分离Am、Cm多是以皮尔逊软硬酸碱理论(HSAB)为指导。在三价镧锕分离中,硬碱,例如含O原子配体更易络合三价镧系(Ln(Ⅲ))而不是次锕系(MA(Ⅲ));软碱,例如含S和N原子的配体更易选择性络合MA(Ⅲ)而不是Ln(Ⅲ)。依此类推,Am(Ⅲ)与Cm(Ⅲ)相比外层轨道电子弥散性更大,半径略大,电荷密度比Cm(Ⅲ)小,相对来说是偏软的酸,与含S和N原子的偏软的碱性配体结合得更好。当前研究较多的配体有双酰胺荚醚类、含N杂环类、氮川类、氨基多羧酸等。
3.1 双酰胺荚醚类
双酰胺荚醚(DGA,或称二甘醇酰胺、烷基-3-氧戊二酰胺,结构示于图1)类萃取剂在超钚元素的分离中研究较多。水溶性N,N,N′,N′-四甲基-3-氧戊二酰胺(TMDGA)可与Am形成ML2配合物,能有效抑制N,N,N′,N′-四辛基-3-氧戊二酰胺(TODGA)对Am(Ⅲ)的萃取[24-25],可用作Am的反萃剂;N,N,N′,N′-四乙基-3-氧戊二酰胺(TEDGA)对金属离子的配位能力弱于TMDGA[26-27],对Cm(Ⅲ)和中、重稀土的配位能力比Am(Ⅲ)强。Chapron等[28]研究了不同取代基的DGA对萃取分离的影响,包括链的长度和支化。
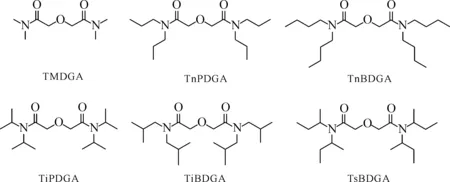
图1 双酰胺荚醚类配体[28]Fig. 1 DGA ligands[28]
TMDGA、N,N,N′,N′-四丙基-3-氧戊二酰胺(TnPDGA)和N,N,N′,N′-四异丙基-3-氧戊二酰胺(TiPDGA)是水相配合剂。研究表明TMDGA、TEDGA和TnPDGA中,二个碳的TEDGA分离因子最高。一个有意思的发现是TEDGA在两相的分配比较复杂。在没有金属离子时TEDGA很少分配到有机相,但是当有阳离子时,TEDGA与阳离子的配合物可以部分进入有机相,与N,N′-二甲基-N,N′-二辛基-2-(2-己基乙氧基)丙二酰胺(DMDOHEMA)和二(2-乙基己基)磷酸(HDEHP)形成加合物。这可能与其较好的Am、Cm分离效果有关联,需要进一步研究。四丁基类似物是作为萃取剂与TODGA进行比较,其中TnBDGA分离能力与萃取能力均比TODGA好。对于萃取剂,β位衍生物会降低Am和Cm的萃取,α位衍生物可以提高Am和Cm的萃取分配比,但是会丧失选择性。
日本东北大学Usuda等[24]合成了水溶性N,N,N′,N′-四乙基-3,6-二氧辛烷二酰胺(DOODA-(C2))和酯溶性N,N,N′,N′-四辛基-3,6-二氧辛烷-二酰胺(DOODA(C8))类萃取剂(图2)。TODGA与DOODA(C2)组合,可以优先萃取Cm,而DOODA-(C8)与TEDGA组合可以优先萃取Am。萃取剂TODGA与DOODA(C8)注入多孔的SiO2-P小球中制成吸附材料。采用TODGA树脂吸附,水相为0.05 mol/L DOODA(C2)的0.1 mol/L HNO3溶液,洗脱液是0.25 mL/min的3 mol/L HNO3,分离因子SF(Am/Cm)为 3.9,但是Am和Cm的吸附容量均较低。采用DOODA(C8)吸附树酯,水相为0.005 mol/L TEDGA的3 mol/L HNO3溶液,吸附容量相对较高,SF(Am/Cm)为2.2。
3.2 氮杂环类
在三价镧锕分离研究中,氮杂环类配体研究较多,主要有吡啶类(BTPs)、联吡啶类(BTBPs)和菲咯啉类(BTPhens)(图3)[29-31]。BTPs与Am可形成ML3配合物,而BTBPs与Am形成ML2配合物[25, 32]。从BTPs到BTBPs,配体的配位点数增加了,构成8~9配位所需要的配体数量减少,这更有利于配合物的稳定。但是联吡啶BTBPs的萃取动力学慢,BTPhens类配体的萃取动力学更快,性能比BTBPs更好[30, 33]。一般认为BTBPs可以通过两个杂环之间的连接键进行翻转,翻转需要克服12 kcal/mol的能垒,所以萃取动力学慢,而BTPhens已经预先构成稳定的半环,萃取动力学较快[32]。BTBPs和BTPhens对Am和Cm均有分离能力。采用CyMe4-BTPhen与水相TEDGA协同萃取,在酸度为0.03~0.5 mol/L时,Am/Cm的分离因子达到5,然而在高酸时Am/Cm的分离因子降到2。文献[34]发现,将BTPhen中1,10-邻菲罗啉的5或5、6位用溴取代后,能增强Am和Eu的分离效果。继而将CyMe4-BTPhen通过反应衍生出溴代和酚羟基取代的配体,结果表明CyMe4-BTPhen本身没有Am/Cm的分离能力,溴取代后在0.1 mol/L HNO3下,SF(Am/Cm)达到7左右,酚羟基取代物的分离因子在1 mol/L HNO3下达到最高,约为5,两个Br取代后,在0.1~3 mol/L HNO3下最大分离因子SF(Am/Cm)约为2[30]。这是通过Am和Cm与配体之间共价作用的细微差异实现分离的,考虑到取代基的位置距离氮杂环较远,这种效应理应是微弱的,但是对于Am和Cm的分离区别却很显著。
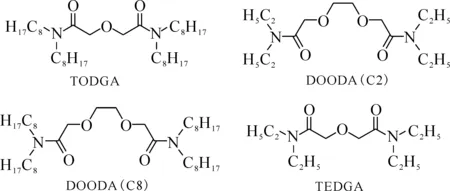
图2 DOODA与DGA类分子组合[24]Fig.2 Combination of DOODA and DGA ligands[24]
Lewis等[35]发现CyMe4-BTPhen配体具有动力学选择性。在非平衡萃取时SF(Am/Cm)最大可以达到7.9。这可能是因为在配体取代Am(Ⅲ)水合物中水分子时,反应比Cm(Ⅲ)的更快造成的,使得萃取Am更快。对这一过程进行数值建模,模型计算与实验数据吻合很好[36],证实Am(Ⅲ)与CyMe4-BTPhen的络合速率是Cm(Ⅲ)的两倍。捷克的t’astn等[37]将萃取剂与聚丙烯腈(PAN)制成固相材料。DMDOHEMA-PAN/TEDGA无法完全分离Am和Cm,TODGA-PAN/(PhSO3H)2-BTPhen则分离效果较好,SF(Cm/Am)可达到约3.5。
水溶性的SO3-Ph-BTBP可以用于反萃Am。有机相采用TODGA共萃取Am、Cm和镧系,然后水相采用SO3-Ph-BTBP反萃取,SF(Cm/Am)约为2.5,并且Eu(Ⅲ)和Am(Ⅲ)的分离因子可以达到1 200。这一过程称之为AmSEL流程[31, 38]。
3.3 氮川类
氮川三乙酰胺萃取剂(图4[26])是近年合成的一类新萃取剂,这类分子中含有软配位N原子,对三价锕系离子有一定的选择性[39-40]。氮川三乙酰胺萃取有如下优点:萃取剂不含S元素和N杂环结构,提高了化学稳定性和辐照稳定性,萃取动力学快,可在10 min内达到萃取平衡。但与BTP和BTBP类含N杂环萃取剂相比,氮川三乙酰胺萃取分离因子不够高。单独的氮川类萃取剂分离Am和Cm时分离因子不高,SF(Am/Cm)约为2,0.5 mol/L NTAamide(C8)在10 mmol/L TEDGA协同作用下,分离因子最高可以达到6.5[26]。氮川类配体的另一个问题是随着酸度的上升,三价锕系离子分配比下降,在HNO3浓度为1 mol/L时,其分配比远低于1。

图3 氮杂环类配体[29-31]Fig.3 Nitrogen-containing heterocyclic ligands[29-31]
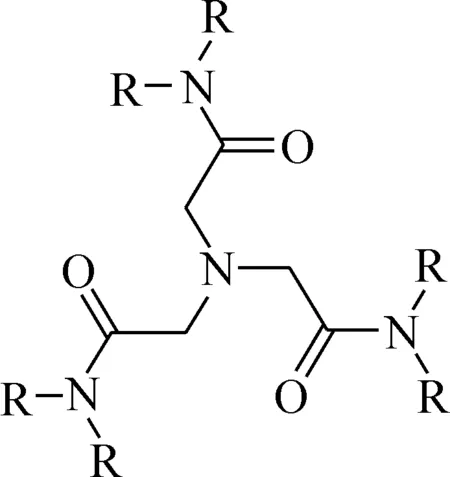
图4 氮川类配体结构[26]Fig.4 Structure of nitrilotriacetamide ligand[26]
3.4 烷基二酰胺胺

ADAAM中对Am萃取分配比最大的是ADAAM(2EH, N(2EH))。通过斜率法得到ADAAM(2EH, N(2EH))萃取Am和Cm均形成1∶1的配合物。随着HNO3浓度从0.1 mol/L增加到1 mol/L,D(Am)和D(Cm)均增加,在1 mol/L时达到最大,并且分离因子也达到最大的5.5,随着HNO3浓度从1 mol/L进一步增加到5 mol/L,D(Am)和D(Cm)又开始下降,这是因为在高H+浓度下,N原子会被H+质子化,造成络合能力下降。当水相中加入TEDGA时,Am和Cm的分配比均会下降,其中Cm的下降更快,最高分离因子可达到40左右,是目前报道的溶剂萃取法分离Am、Cm最高分离因子。对其进行混合澄清槽连续萃取实验。进料为含有Am和Cm的1.5 mol/L HNO3溶液,有机相为0.25 mol/L ADAAM/正十二烷,洗涤液为1.5 mol/L HNO3,反萃液为0.05 mol/L HNO3。经过8级萃取、8级洗涤和16级反萃,回收了99.8%的Am,其中含有的Cm少于9.6%。从ADS嬗变所需Am的燃料来说,是满足需要的。而Am(Ⅲ)基本完全从溶液中去除,Cm中含有的Am非常低[43]。只通过一种萃取剂就实现了Am和Cm的很好分离,是非常有前景的分离体系,仍有待热实验的考验。
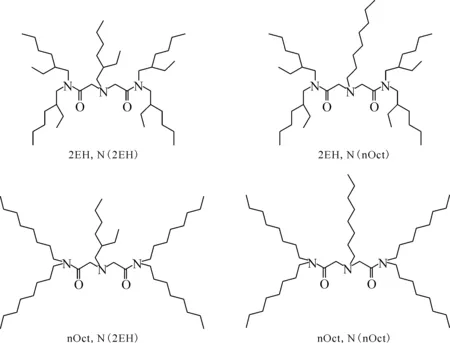
图5 ADAAM类萃取剂[42]Fig.5 Diamide amine extractants[42]
通过密度泛函理论(DFT)计算可将HSAB规则通过成键布居分析来表达,即络合时金属f轨道与配位原子共价作用的程度。金属5f轨道与配位原子轨道混合的程度和轨道类型是决定选择性的关键因素,而不是定域化5f轨道能量[44]。Am-TMDGA的长度比Cm-TMDGA长,但是Am-ADAAM的长度比Cm-ADAAM短,表明Am-ADAAM比Am-TMDGA有更强的共价性。其他研究也表明,Am-N键比Cm-N键强[45],而Am-O键比Cm-O键强度要弱[46]。
3.5 氨基多羧酸类
N,N,N′,N′-四[(6-羧基吡啶-2-基)甲基]乙二胺 (H4TPAEN)最早由Chatterton等[47]合成,用作镧系离子的有机生色团。Gracia等[48]合成了10种H4TPAEN的衍生物(图6)。在这些配体中,H4TPAEN对Am和Cm的分离最佳。L2和L3对于Am的亲和能力也比较强,但是分离因子较低。L1在水中的溶解度略有提高,并且在反萃TODGA中的Am时操作性能较好,但是相较于H4TPAEN,L1与Am的亲和能力略有下降,使用5 mmol/L配体,在pH=1.5时,SF(Cm/Am)可达到3.2。法国CEA为了简化开发的EXAm流程,采用TODGA作为萃取剂,稀释剂是氢化四聚丙烯烃(TPH)+5%正辛醇,将Am和Cm一同萃入有机相,然后采用H4TPAEN反萃Am[49]。TPAEN配体主要不足是在酸性溶液中溶解度低,最高约10 mmol/L,并且反萃过程pH值约为1。
Boubals等[50]研究了H4TPAEN与Am、Cm络合机理。采用X射线吸收精细结构谱(EXAFS)研究了Am与TPAEN的结构(图7)与计量比。并用镧系元素研究了络合稳定常数。Am与TPAEN
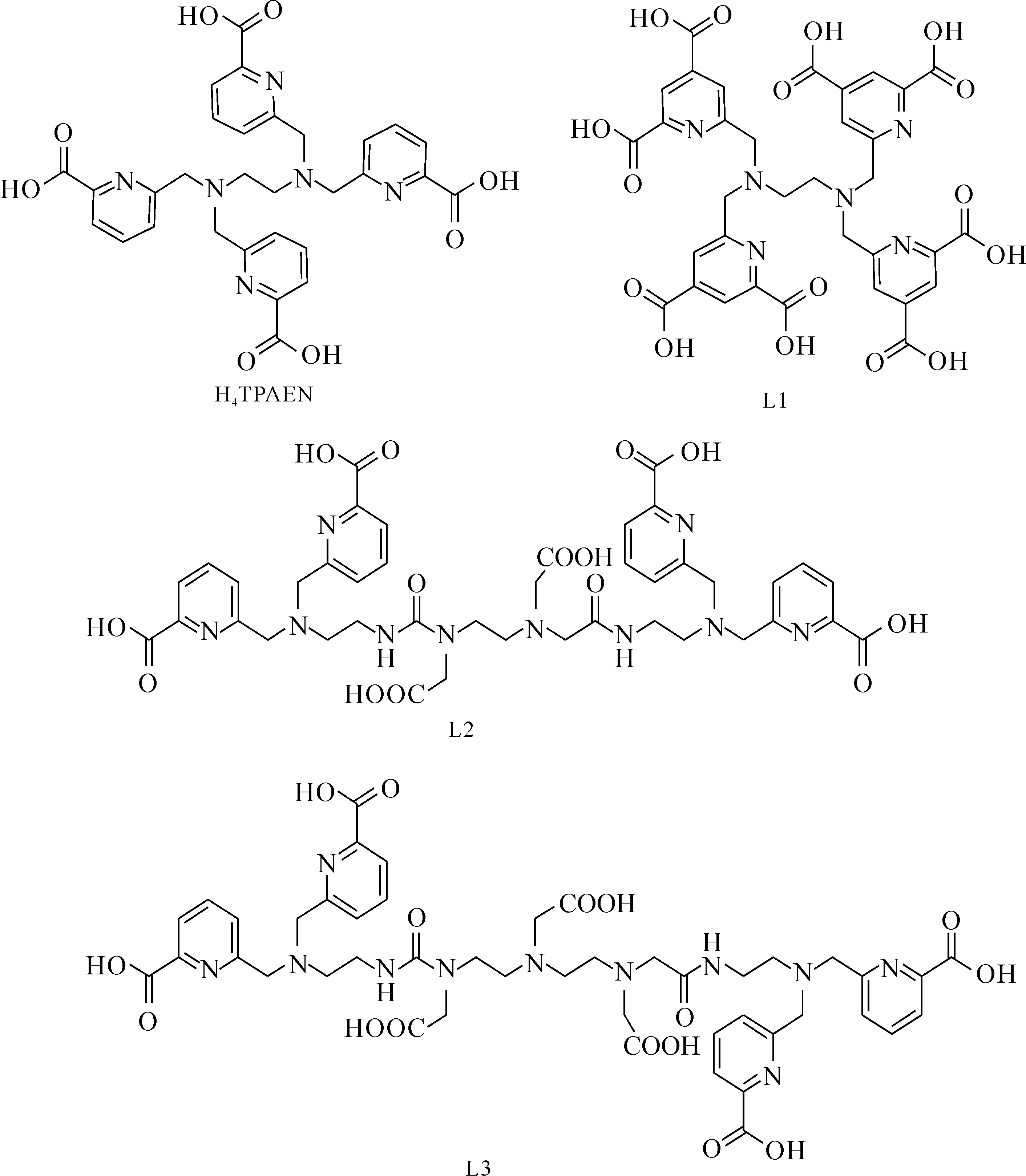
图6 H4TPAEN及其衍生物[48]Fig.6 H4TPAEN and its derivatives[48]
通过10个原子配位,即6个N原子和4个氧原子。这比Am的水合物(通常是8~9个水)的配位数要高,因此Am与配位原子的距离也比9配位的配合物要长一些。尽管配位点很多,但是键长较长,因此TPAEN的分离效果并不突出。TPAEN与Am和Cm的络合稳定常数(lgβ)分别为4.5±0.2和4.3±0.2,络合选择性ΔΔG(Am/Cm)=-1.1 kJ/mol。在TODGA/TPAEN协同体系,ΔΔG约为3 kJ/mol,Am和Cm分离因子约达到3.5~4.0。Huang等[45]对TPAEN分离Am、Cm进行了计算,Am与Cm的配合物结构几乎完全相同,只是金属与N原子的距离有很小的差别;在金属与N和O配位时,主要还是静电相互作用,但是也发现有微弱的金属与N的共价成键。
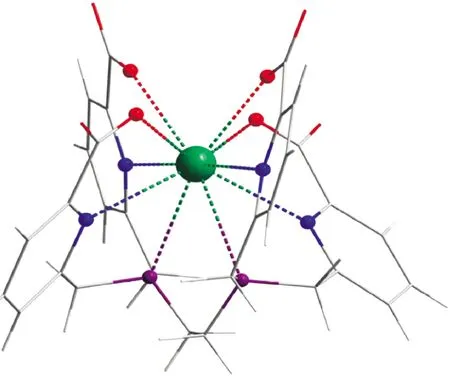
图7 Am-TPANE配合物可能的构型[50]Fig.7 Possible configuration of Am-TPANE complex[50]
3.6 18-冠-6
Cm(Ⅲ)只比Am(Ⅲ)的半径小一点,对于8配位,其半径分别是1.096 Å与1.106 Å。因此Cm(Ⅲ)具有相对较大的电荷密度。通常Cm与配体络合时吉布斯自由能往往比 Am的配体有0.5~2 kJ/mol的优势。美国阿贡国家实验室(ANL)的Jensen等[51]采用N,N′-双[(6-羧基-2-吡啶基)甲基]-1,10-二氮杂18-冠醚-6(H2bp18c6)分离Am和Cm,可优先萃取Am,分离因子为4.1(ΔΔG=3.5 kJ/mol)。因为该配体中也含有软配位原子N,无法完全将其分离效果归结于尺寸效应。H2bp18c6配体与配合物优化构型示于图8[51]。
此外也有通过萃取色层或络合后电场迁移等手段进行分离。将金属离子与2-羟基异丁酸(HIBA)形成配合物,因为离子半径不同,有效电荷密度也存在差异,在电场下迁移速率不一样[52]。德国Karlsruhe联合研究中采用强阳离子交换HPLC树脂,分离毫克级242Cm[53]。Hanson等[54]报道了一种光化学反应氧化Cm(Ⅲ)配合物的方法,并通过Cm(Ⅳ)配合物的配体交换反应,实现Am与Cm的分离。
4 典型流程中Am与Cm的分离
SANEX(selective actinide extraction)流程采用CyMe4-BTBP和DMDOHEMA萃取体系,对Am和Cm的回收率均比较高, 达到99%,但是不进行Am和Cm相互分离[55]。
SESAME(selective extraction of americium by electrochemical methods)是基于氧化法分离Am、Cm的流程,采用电化学方法将Am氧化到Am(Ⅵ),并用杂多阴离子络合以稳定Am(Ⅵ),然后采用磷酸三丁酯(TBP)萃取Am(Ⅵ)。2001年在法国Atalante进行了真实料液热实验。在高放射性料液中,由于辐解产物的影响,很难使萃取到有机相的Am(Ⅵ)保持价态不变,造成Am的回收率较低(78%),Cm的回收率为88%,并未达到预期效果[55]。
LUCA(lanthaniden und curium americum separation) 流程是专门为了Am、Cm分离而设计的流程(图9)[56],该流程采用双(4-氯苯基)-二硫代次膦酸(ClPh)2PSSH)以及磷酸三(-2-乙基己基)酯(TEHP)协同萃取。该流程可获得很高的Am对镧系的选择性,SF(Am/Eu)>3 000,Am和Cm的分离因子也很高,SF(Am/Cm)可达到6~10,这是因为采用了较软的含S络合剂(ClPh)2PSSH。2008年对LUCA流程进行了台架试验[57]。进料组成为模拟料,包括了Am、Cm、Cf、Eu,0.13 mol/L HNO3,0.5 mol/L NaNO3。采用我国1 cm离心萃取器,通过8级萃取、8级洗涤和8级反萃,结果如预期的,只有Am被定量萃取,并且用0.7 mol/L HNO3反萃,回收率可达到99.8%。废液中Cm(Ⅲ)量>99.5%(相对原始溶液的量),Cf量>99.9%,Eu量>99.9%,含有的Am(Ⅲ)量<0.08%。反萃后的Am只含有0.47%的Cm(Ⅲ),DF(Am/Cm)达到214。DF(Am/Eu)高达7 693。萃取剂(ClPh)2PSSH在辐照剂量达到0.5 MGy时也是稳定的,主要问题是在高酸时不稳定,例如在反萃时,酸度>0.5 mol/L,配体会有相当程度的降解。需要用0.08 mol/L HNO3低酸进料, 在放射性较强的情况下还需防止HNO2对络合剂的破坏, 须使用HNO2清扫剂,或者在反萃时换成HCl介质。

图8 H2bp18c6配体与配合物优化构型[51]Fig.8 Configurations of H2bp18c6 ligand and complex[51]
EXAm (extraction of americium process) 流程由法国CEA开发,目前研究较为活跃,其流程如图10所示[3, 57-58]。近期Miguirditchian等[3]详细介绍了法国在高放废液处理方面的政策过程、20年的次锕系元素分离研究,特别是EXAm流程的发展。该流程采用DMDOHEMA+HDEHP为萃取剂,其最重要的特点是水相采用TEDGA络合剂掩蔽。TEDGA是含O原子的水溶性配体,可以优先络合偏硬的Cm(Ⅲ),加入TEDGA后,SF(Am/Cm)从1.6升高到2.5。2010年采用真实PUREX高放废液进行热实验,2015年采用高放废液浓缩液进行实验,地点在法国Atalante的CBP屏蔽线,共回收了2.4 g Am,并且各级浓度分布的模拟计算与实验结果匹配很好[59]。采用68级混合澄清槽(32级用于Am的萃取和Am/Cm分离),对Am的回收率为98.3%,对Cm的去污因子DF(Am/Cm)达到500,对镧系去污因子DF(Am/Nd)达到340。由于该体系SF(Am/Cm)并不突出,即使加入TEDGA后有所提升,所需的萃取级数仍较多,流程较复杂。
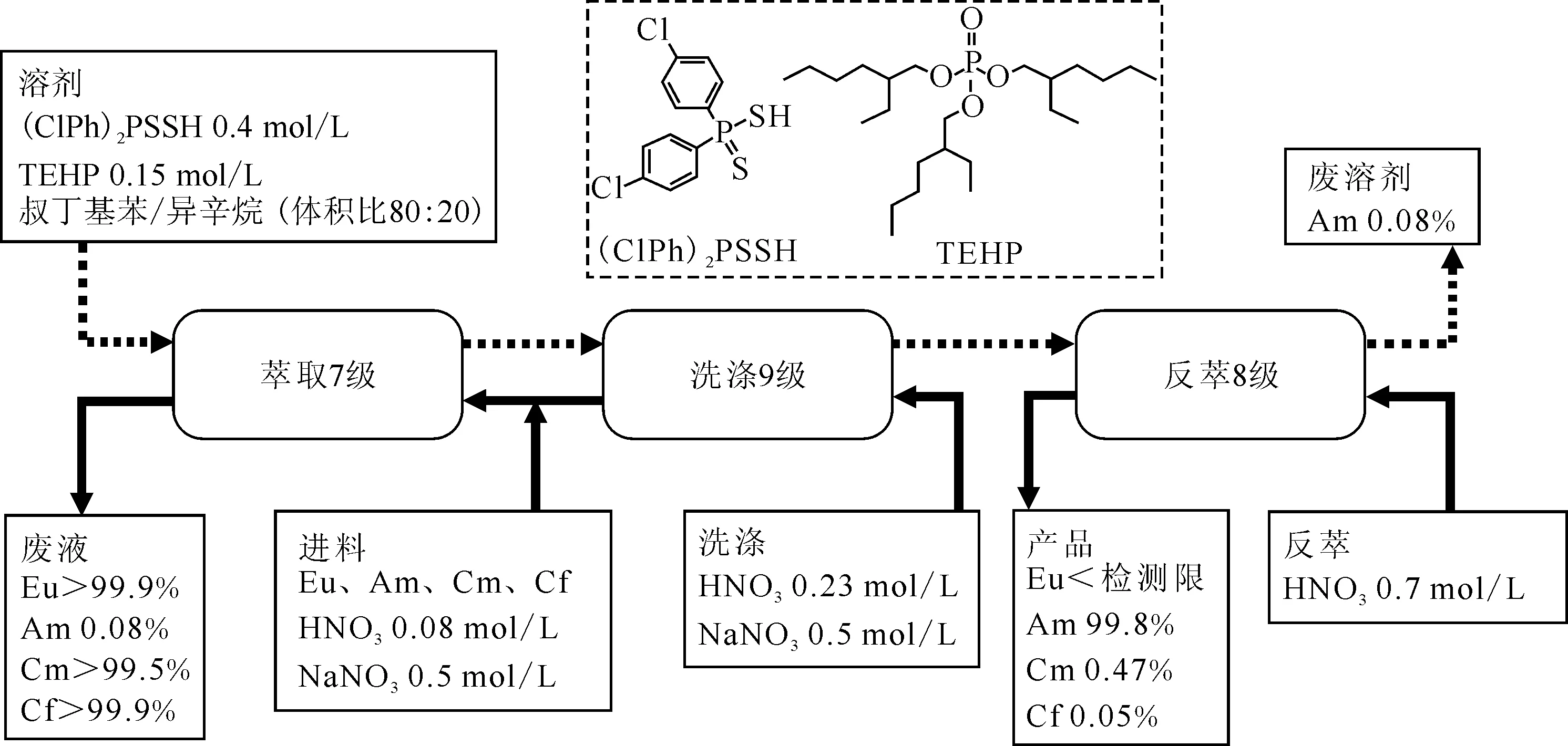
虚线表示有机组,实线表示水相图9 LUCA流程及所用试剂[56]Fig.9 LUCA process[56]
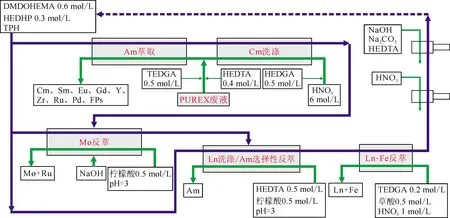
虚线表示回收复用,蓝色实线表示有机相,绿色实线表示水相图10 EXAm流程示意图[3]Fig.10 EXAm process[3]
为此在欧洲SACSESS项目中又设计了简化的分离流程,主要是减少了洗涤级数以提高流程的紧凑性。首先使用TODGA为萃取剂,共萃取Am(Ⅲ)和Cm(Ⅲ),选择TODGA是因为即使在低酸度下,Am(Ⅲ)的分配比也较高,并且含O配体会萃取偏硬的Cm(Ⅲ)比Am(Ⅲ)稍多一点,这在Am反萃时可以提高Cm/Am的选择性。然后第二步采用水相络合剂H4TPAEN在较低酸度下(pH=1)选择性反萃Am,而保持Cm在有机相中仍与TODGA结合。该体系中Cm、Am的分离因子可达到3.5~4。H4TPAEN可以与Am和Cm形成1∶1配合物,lgβ(Am)=4.5±0.2,lgβ(Cm)=4.3±0.2。Am的H4TPAEN配合物在pH=1时仍是稳定的,这就意味着反萃可以在较低酸度下操作,不需要引入缓冲剂到水相。这一流程的主要缺点是H4TPAEN在水溶液中溶解度较低,最高为10 mmol/L,因此有沉淀的风险,仍需对H4TPAEN的取代基进行优化,以提高溶解度。
5 国内研究概述
国内相关研究主要是在20世纪80年代。研究涉及离子交换法、溶剂萃取法以及相关的分析技术。西北核技术研究所研究了Am和Cm与三乙四胺六乙酸(TTHA)、吡啶2,6-二羧酸、2-羟基-2,4-二甲基戊酸等配体的络合稳定常数[60-63]。兰州大学陈励权等[64]研究了采用加压离子交换排代法提取Am、Cm和钷(Pm)的研究, 使用二乙基三胺五乙酸(DTPA)排代提取Am和Cm,氨基三乙酸(NTA)排代提取Pm。陈耀中等[65]实验了高压离子色层分离Am和Cm,采用了聚苯乙烯三甲胺型阴离子树脂,可以很好地分离Am和Cm,Cm产品纯度大于99.9%。张力争等[66]采用硝酸甲基三烷基铵萃取Am和Cm,分离因子可以达到3,当水相中加入DTPA时,分离因子可提高至4.2。清华大学采用Cyanex301对Am、Cm分离进行了研究,单次分离因子为3,并进行了串级模拟实验[67]。中国原子能科学研究院朱荣保等[68]建立了超钚元素分离流线监测系统,吴克明等[69]从数克辐照的241Am中提取百居里的242Cm,Am、Cm的分离采用高压阳离子交换树脂,淋洗液为0.04 mol/L α-羟基异丁酸,过程采用在线监测,说明高压离子交换分离Am和Cm是可行的。
6 结论与展望
离子交换法分离Am、Cm目前仍在使用,多用于辐照Am靶中提取Cm,因为体系组成相对单纯,采用离子交换法分离Am、Cm通常可满足提取需要。当前研究较多的则是基于Am(Ⅲ)的氧化或通过Am(Ⅲ)和Cm(Ⅲ)与配体络合的强弱差异来实现分离。Am的氧化法分离中容易将Am定量氧化到Am(Ⅵ),但是在溶剂萃取或离子交换时,常因为试剂纯度、辐解产物等影响,造成高价态Am的迅速还原。将Am氧化到相对稳定的Am(Ⅴ),是氧化法分离Am、Cm研究的新思路。因为国外对高放废液中次锕系元素的管理愈发关注,采用萃取法对Am、Cm进行分离是研究热点。萃取法分离Am、Cm的基本策略是采用“推拉效应”,有机相萃取剂倾向于萃取Am(Ⅲ)或Cm(Ⅲ),水相络合剂倾向于络合Cm(Ⅲ)或Am(Ⅲ),从而放大分离效果。近年不乏新型萃取剂的设计、实验与量化计算的研究报道,提出了分离效果更好的萃取剂及萃取体系,并力图从理论计算上探索Am、Cm配合物结构、结合能的微弱差异与分离性能的关系。我国坚持闭式燃料循环路线,需对高放废液中的Am和Cm进行妥善处理,另外因为超钚元素生产的需求,需要开展Am、Cm分离基础研究,发展Am、Cm分离技术。
