Molecular mechanisms of viral hepatitis induced hepatocellular carcinoma
2020-12-10SimmonesouzaKeithCKLauCarlaCoffinTrusharPatel
Simmone D'souza, Keith CK Lau, Carla S Coffin, Trushar R Patel
Abstract Chronic infection with viral hepatitis affects half a billion individuals worldwide and can lead to cirrhosis, cancer, and liver failure. Liver cancer is the third leading cause of cancer-associated mortality, of which hepatocellular carcinoma (HCC) represents 90% of all primary liver cancers. Solid tumors like HCC are complex and have heterogeneous tumor genomic profiles contributing to complexity in diagnosis and management. Chronic infection with hepatitis B virus (HBV), hepatitis delta virus (HDV), and hepatitis C virus (HCV) are the greatest etiological risk factors for HCC. Due to the significant role of chronic viral infection in HCC development, it is important to investigate direct (viral associated) and indirect (immune-associated) mechanisms involved in the pathogenesis of HCC. Common mechanisms used by HBV, HCV, and HDV that drive hepatocarcinogenesis include persistent liver inflammation with an impaired antiviral immune response, immune and viral protein-mediated oxidative stress, and deregulation of cellular signaling pathways by viral proteins. DNA integration to promote genome instability is a feature of HBV infection, and metabolic reprogramming leading to steatosis is driven by HCV infection. The current review aims to provide a brief overview of HBV, HCV and HDV molecular biology, and highlight specific viral-associated oncogenic mechanisms and common molecular pathways deregulated in HCC, and current as well as emerging treatments for HCC.
Key Words: Chronic viral infection; Hallmarks of cancer; Hepatocellular carcinoma; Hepatitis B virus; Hepatitis C virus; Hepatitis delta virus co-infection; Molecular mechanisms; Viral hepatitis
INTRODUCTION
Epidemiology of viral hepatitis associated hepatocellular carcinoma
Liver cancer is the third leading cause of cancer-associated mortality (781631 people/year), despite being ranked seventh on global incidence (841080 people/year)[1]. Approximately 12% of all cancer cases globally arise from chronic infections with bloodborne oncogenic viral pathogens including hepatitis B virus (HBV), hepatitis C virus (HCV), and hepatitis delta virus (HDV)[2]. Although incidence in the majority of cancers has decreased, primary liver cancer incidence is the fastestgrowing cancer with regards to incidence and mortality[3]. Hepatocellular carcinoma (HCC) represents 90% of all liver cancer cases and the risk factors are well defined: Viral infection with HBV, (54% of all HCCs) and/or HCV (31% of all HCCs), cirrhosis (80% of all HCCs), high alcohol consumption, obesity, genetic disorders such as hemochromatosis, exposure to aflatoxins, sex (male) and older age (50+)[4-7].
Virus-induced HCC is present worldwide, however, there are considerable differences in populations that develop HBV or HDV induced HCCvsHCV induced HCC. HBV and HDV associated HCC is more common in low and middle-human development index countries, while HCV induced HCC is more common in high and very high-human development index[2]. Chronic hepatitis B (CHB) infection affects around 257 million people worldwide, of which 48-60 million people are co-infected with HDV and an estimated 2.6 million are co-infected with HCV[8-10]. Exposure to infected blood/bodily fluids is the primary mode of transmission for HBV and HBV/HDV, with majority of exposures occurring from mother to child during birth or early years of life. Unvaccinated neonates and children who have been exposed to HBV have > 95% risk of developing chronic disease, while infection during adulthood results in < 2% chance of developing chronic disease[11]. HBV/HDV co-infection have the highest mortality rate (20%) associated with any viral hepatitis infection and most severe liver disease (i.e.acute liver failure, cirrhosis within 5 years, and HCC within 10 years)[10,12,13].HCV has established chronic infection in 70 million people primarily through horizontal blood-borne transmission routes such as intravenous drug use, needle pricks, unscreened blood transfusions, and high-risk sexual practices[11]. In comparison to HBV or HCV mono-infection, individuals who are co-infected with HBV/HCV have increased rates of HCC development. Overall, viral etiologies represent approximately 80% of all HCC related cases, highlighting the importance of investigating the role of these viruses in the development of liver cancer.
Preventative measures against HBV and HDV induced liver cancer include birthdose vaccinations, hepatitis B immunoglobulin treatment for children born to infected mothers as well as treatment of mothers with high HBV viral load with nucleos(t)ide inhibitors in the third trimester[14]. For those individuals who are already chronic carriers of HBV/HDV, there is no virological cure; however, treatment with nucleos(t)ide analogs can lower the risk of HCC development[15]. There is no protective vaccine available for HCV, but there are effective direct-acting antivirals that can cure > 90% of chronic carriers. Those who have a sustained virological response from direct-acting antiviral treatment have a significantly lower risk of HCC development if cirrhosis is absent[16]. Although there are treatment options to lower the risk of HCC in those who have chronic viral hepatitis infection, globally many individuals are unaware of their status, lack access to testing, and effective treatment.
In this review article, we discuss the molecular biology of HBV, HCV, and HDV, common features associated with virus-induced cancers, viral oncogenic mechanisms leading to HCC relating to the hallmarks of cancer, common molecular pathways deregulated in HCC, and current as well as emerging treatments for HCC.
OVERVIEW OF VIRAL LIFE CYCLES
Hepatitis B virus life cycle
The HBV is a member of theHepadnaviridaefamily, which has a cellular tropism for hepatocytes, but has also been detected in extra-hepatic reservoirs such as the lymphoid cells (i.e.peripheral blood mononuclear cells)[17-20]. HBV has a compact 3.2 kb partially double-stranded relaxed circular DNA genome (rcDNA) containing four overlapping open reading frames: Pre-S/S, X, P, and pre-C/C, which are under the transcription control of the pre-S1 promoter, pre-S2/S promoter, enhancer I/X and enhancer II/basal core promoter[21]. The viral protein products include three surface proteins (large/pre-S1, middle/pre-S2, and small/S - also known as HBsAg), the core antigen (HBcAg), the excreted “e” antigen (HBeAg), the viral polymerase (which has reverse transcriptase, DNA polymerase, and RNaseH activity), and the X protein (HBx) that plays an essential role in HBV pathogenesis and viral transcription[21]. Upon viral attachment of the envelope HBV preS1 protein to the sodium taurocholate cotransporting polypeptide receptor, the virus is endocytosed (Figure 1). The nucleocapsid is transportedviamicrotubules from the cytoplasm to the nucleus where the rcDNA is converted to covalently closed circular DNA (cccDNA)[22,23]. The cccDNA associates with histone and non-histone proteins which form a viral minichromosome that persists in the hepatocyte to serve as the template for transcription of pregenomic RNA (pgRNA) and subgenomic RNAs by host RNA polymerase II[24]. The exported pgRNA and subgenomic transcripts are translated to produce the core protein, viral envelope surface proteins, HBeAg, polymerase, and X proteins. In addition, the pgRNA transcript is packaged by the capsid proteins and reverse transcribed by the viral polymerase into rcDNA. The newly packaged rcDNA can either localize back to the nucleus to replenish the cellular cccDNA population or gain their coat through the endoplasmic reticulum (ER)/Golgi and proceed to bud out of the to infect other cells[25]. Current nucleos(t)ide antivirals target the viral reverse transcriptase to produce aberrant transcripts that cannot produce infectious virions. Additional details about the lifecycle and host-transcription factors/proteins required for HBV replication are included in our previous article by Turtonet al[26].
Hepatitis Delta virus life cycle
HDV is the smallest human infecting virus and the sole member of theDeltavirusgenus[27]. HDV is characterized as a “satellite” or “defective” virus as it is dependent on HBV co-infection for viral assembly and persistence. HDV has an approximate 1.7 kb circular, single-stranded, negative-sense RNA genome that encodes for a single protein of two isoforms: The small and large delta antigens (S-HDAg and L-HDAg, respectively)[28]. Viral entry (Figure 2) occurs similarly to HBV due to HDV’s co-opted use of the envelope HBsAg protein[29]. Following viral entry, HDV uncoats in the cytoplasm and the ribonucleoprotein complex consisting of the HDV viral genome and HDAg complex is imported into the nucleus[30]. Rolling-circle replication occurs in the nucleolus using the host RNA polymerase II to produce antigenomic positive sense HDV RNA that serves as a template for genomic HDV RNA synthesis and protein production[31]. The antigenome can be edited byhost protein adenosine deaminase act- ing onRNA1 (ADAR1) to change adenine to inosine in the UAG stop-codon to produce the L-HDAg. The edited and non-edited antigenomes are then linearized by the HDV associated ribozyme, exported to the cytoplasm, and translated to HDV antigens. The non-edited transcript produces S-HDAg (24 kDa) and the transcript modified by ADAR1 produces the L-HDAg (27 kDa)[32]. Following extensive posttranslational modifications, the viral antigens associate with the HDV RNA in the cytoplasm to form the ribonucleoprotein complex. The ribonucleoprotein is trafficked through the ER and Golgi apparatus where it co-opts the HBsAg envelope produced by HBV, and then buds out of the cell[33].
Hepatitis C virus life cycle

Figure 1 Hepatitis B virus life cycle. Viral entry is mediated by low-affinity binding of the Pre-S1 protein to the heparin sulfate proteoglycan receptor, followed by binding to the sodium-taurocholate co-transporting polypeptide to facilitate entry. The nucleocapsid is transported from the cytoplasm to the nucleus where the relaxed circular DNA (rcDNA) genome is converted into the persistent covalently closed circular DNA (cccDNA) form. Viral mRNA is then transcribed from the cccDNA genome and translated at the rough endoplasmic reticulum. The greater than genome length pregenomic RNA is transported to the cytoplasm, encapsidated by the hepatitis B virus core protein and reverse transcribed by the hepatitis B virus polymerase to produce rcDNA or double-stranded linear DNA. The core particles can then obtain their envelope proteins at the endoplasmic reticulum to be excreted out of the cell, or the core particles containing double-stranded linear DNA can relocate into the nucleus and integrate into the host genome, and the rcDNA can be recycled intracellularly to replenish the cccDNA pool.
The HCV is part of theFlaviviridaefamily and the genusHepacivirus.HCV is an enveloped, positive-sense, 9.6 kb single-stranded RNA virus with highly conserved 5’ and 3’ untranslated regions[34]. HCV primarily infects hepatocytes due to the expression of essential entry receptors and liver-specific cellular host factors (miRNA-122) required for viral replication[35]. However, extrahepatic manifestations have been observed in peripheral blood mononuclear cells, epithelial cells, kidneys and in the peripheral nervous system[36]. Through complex mechanisms, HCV particles interact with several receptors (see[37]for details) to induce conformational changes and proceeds to enter the cell (Figure 3)viaclathrin-mediated endocytosis[37]. Endosomal acidification causes the fusion of the viral envelope to the endosome membrane, disassociation of the viral core, and release of the HCV RNA genome into the cytoplasm[37]. In the ER the viral RNA is replicated and translated from a single open reading frame using the 5’ untranslated regions internal ribosomal entry site. The translated product is an approximately 3000 amino acid polyprotein precursor that is cleaved by host and viral proteases to form ten proteins[38]. There are three structural proteins - core, E1 and E2 - and seven non-structural proteins p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B that have roles in polyprotein cleavage, viral replication, assembly, and release[39]. More recently, two isoforms of the core protein, known as the “mini-core” were discovered to be translated from an alternative open reading frame at amino acids 70 and 91 which preserve the c-terminal portion of the mature p21 core nucleocapsid but lack the N-terminus. The function of these mini-core proteins has yet to be elucidated, however, mutations in amino acid positions 70 and 91 are associated with increased risk of HCC, insulin resistance, and failure of interferon treatments[40,41]. Following viral replication and protein translation, the core protein assembles in the ER on a lipid droplet and recruits HCV viral RNA which is subsequently encapsidated[42]. The viral nucleocapsid is then processed through the ER-lumen and Golgi apparatus for maturation and the HCV particle associated with very low-density lipoproteins are released from the plasma membrane[38,43,44].

Figure 2 Hepatitis delta virus lifecycle. Viral entry is mediated (like hepatitis B virus) by low-affinity binding of the Pre-S1 protein to the heparin sulfate proteoglycan receptor, followed by binding to the sodium-taurocholate co-transporting polypeptide to facilitate entry. Following uncoating, the ribonucleoprotein (RNP) complex consisting of negative-sense single-stranded RNA genome plus the small and large hepatitis delta virus (HDV) antigens (L-HDAg/S-HDAg) are transported to the nucleus. Within the nucleolus, HDV RNA is replicated using a double rolling circle amplification to form the positive-sense anti-genomic RNA and more genomic RNA. From the amplification process, the genomic RNA is transported out of the nucleolus and into the nucleus where it can be transcribed to produce the S-HDAg transcript or undergo A to I editing by ADARI to produce the L-HDAg RNA. Once the RNA transcripts are exported out of the nucleus, translation machinery produces the S-HDAg and L-HDAg which associate with the genomic HDV RNA to produce the RNP complex. The RNP complex passes through the endoplasmic reticulum and Golgi apparatus to obtain its coat and are then released out of the cell to infect neighboring hepatocytes. ER: Endoplasmic reticulum; NTCP: Sodiumtaurocholate co-transporting polypeptide; HSPG: Heparin sulfate proteoglycan receptor; RNP: Ribonucleoprotein.
Overview of mechanisms driving HCC development with infection by HBV, HCV, HDV
HBV, HCV, and HDV use several mechanisms to co-opt the infected cells which may unintentionally lead to HCC development. Commonly used mechanisms between all three viruses include: (1) Persistent liver inflammation and immune-mediated oxidative stress damage from a chronic viral infection; (2) Intracellular oxidative stress damage induced by viral proteins; and (3) Deregulation of cell signaling pathways by viral proteins (e.g. HBx, L-HDAg, S-HDAg, HCV core, NS3, and NS5A/B). HBV is the only hepatotropic DNA virus that also uses viral DNA integration to induce genome instability, which can lead to the creation of fusion gene products, and altered expression of oncogenes or tumor suppressors. In addition, HCV facilitates metabolic reprogramming leading to steatosis, which aids in the progression of fibrosis and HCC.
General traits of oncogenic viruses

Figure 3 Hepatitis C virus life cycle. Hepatitis C virus entry is facilitated by a variety of receptors and signaling pathways (described in[37]). Upon viral entry, the positive sense RNA genome is released into the cytoplasm from endosomal acidification. The viral RNA undergoes replication and translation at the rough endoplasmic reticulum to produce a single polyprotein chain at the endoplasmic reticulum membrane that is cleaved by viral and host proteases into 10 different viral proteins (structural and non-structural). The virus particles are assembled on lipid droplets and associate with very-low-density lipoproteins which mature at the Golgi apparatus and are released via the secretory pathway. HSPG: Heparin sulfate proteoglycan receptor; LD: Lipid droplets; VLDL: Very-low-density lipoproteins.
There are several viral traits that are common to human oncogenic viruses[45,46]: (1) Oncoviruses are ubiquitous in the environment and infection alone is not sufficient for cancer development. Although chronic infection with HBV/HCV/HDV results in higher rates of HCC development, not all persistently infected individuals develop liver cancer. Thus, this observation would suggest that the viruses by themselves are insufficient for cancer development. (2) Virally induced cancers are biological accidents as tumor formation is not an intentional outcome of viral infection. (3) Viral cancers appear in the context of persistent infections and occur many years to decades after initial exposure. Hepatic viruses have co-evolved with their hosts and hence, have evolved effective immune evasion strategies to establish long-term infection such as expression of viral proteins that interfere with innate interferon responses, inflammation, and adaptive immunity. (4) Most viral remnants within a tumor are non-infectious and tumors are non-permissive for viral replication. Active virion production is typically absent in transformed tumor cells. (5) All virally induced cancers have non-infectious co-factors that influence tumorigenesis. Host factors such as age, sex, genetics, environmental factors, and immunodeficiencies are associated with viral hepatitis-related HCC. (6) The immune system can play a deleterious or protective role, with some virus-associated cancers increasing with immunosuppression and others appearing during chronic inflammation. In the context of viral hepatitis induced HCC, the host antiviral immune response is unable to eliminate virally infected cells and instead causes immune-mediated damage. This phenomenon is evident in chronic infections where bouts of repeated hepatitis caused by the inflammation-necrosis-proliferation cycle leads to the production of reactive oxygen species (ROS) that promote genetic mutations, fibrosis, cirrhosis, and HCC.
The features of oncogenic viruses described above reflect the multifactored nature of virus-induced hepatocarcinogenesis. Human oncovirus infection alone is insufficient to directly drive cancer and viral infection provides only a portion of the oncogenic alterations. The combination of viral factors and other factors (i.e. host, environment, time) is generally required for the development of cancer[45]. The hallmarks of cancer outline developed by Hanahan and Weinberg[47]deconstruct the specifics of cellular deregulation into factors that contribute to cancer development[47]. This outline also explains the reliance on time progression to accumulate oncogenic mutations and the multistep nature of acquiring various hallmarks that eventually lead to cancer. By applying this system to viral-induced cancers, we can better understand how alterations in cellular processes induced by hepatitis viruses contribute to HCC development (depicted in Figure 4 and Figure 5).
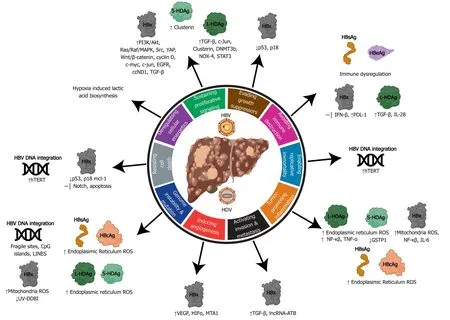
Figure 4 Relating the hallmarks of cancer to the molecular mechanisms of hepatitis B virus and delta virus hepatocarcinogenesis. Hepatitis B virus can activate all ten hallmarks of cancer using viral proteins (HBx, HBsAg, HBeAg, HBcAg) and DNA integration. Hepatitis delta virus has been linked to four hallmarks, primarily through molecular mechanisms manipulated by the large and small hepatitis delta virus antigens (L-HDAg and S-HDAg). HBV: Hepatitis B virus; HDV: Hepatitis delta virus; ER: Endoplasmic reticulum; ROS: Reactive oxygen species.
Specific viral factors affecting HCC development
Viral genotypes vary across the globe and play an important role in virus treatment response and assessing HCC risk. HBV has ten genotypes (A-J) which have a genetic divergence of > 8%. The HBV genotypes associated with the highest risk of HCC development are genotype C > B > F > D > A[7,48]. Some studies suggest that individuals infected with either HBV genotype B or C that have T1762/A1764 basal core promoter mutations have a higher risk of HCC development in younger individuals (< 50 years old) without cirrhosis[49]. In HBV genotype C infections, mutations/deletions in the preS region, enhancer II at position C1653T, and/or T173V in the basal core promoter can predict the development of HCC in 80% of cases[50]. Moreover, genotypes A and B have a better response to peg-IFN-α therapies, while there are no genotypic preferences for nucleos(t)ide analog treatments[51].
HCV has 6 major genotypes (1-6) that have a genetic divergence of 31%-35%. With HCV, studies linking genotype to the risk of developing HCC have inconsistent findings[7]. However more recently, a large cohort study of United States veteran concluded that HCV genotype 3 infections had an 80% higher risk of HCC development compared to genotype 1[52]. In a southeast Asian cohort, HCV genotype 6 is also associated with an increased is of HCC development[53]. With currently approved direct-acting antiviral treatments for all HCV genotypes, sustained virological response is observed in > 90% of treated individuals and reduces HCC risk in individuals without cirrhosis[54].
There are eight different HDV genotypes (1-8), which have a large genetic divergence ranging from 20%-40%[55]. There has not been a significant amount of research conducted to elucidate the effects of HDV genotypes on clinical outcomes. One study concluded that genotype 1 is associated with worse clinical disease including HCC than genotype 2[56]. Moreover, clinical outcomes of the disease are potentially regulated by both HBV and HDV genotypes. Due to the reliance of HDV on HBV co-infection, the only treatment option for HDV infection is peg-IFN-α, which is poorly tolerated and has < 30% response rate, highlighting the urgent need for improved therapies[57].

Figure 5 Relating the hallmarks of cancer to the molecular mechanisms of hepatitis C virus hepatocarcinogenesis. Hepatitis C virus uses its RNA genome and many viral associated structural and non-structural proteins to alter cellular pathways to influence all ten hallmarks of cancer. HCV: Hepatitis C virus.
CHRONIC INFLAMMATION-MEDIATED BY VIRAL HEPATITIS
Non-resolving inflammation is a hallmark of cancer that significantly contributes to the development and progression of HCC[47]. Approximately 80% of HCC cases arise from hepatocyte injury and chronic inflammation resulting in cirrhosis[6,58]. HCC in chronic hepatitis B, C, or HBV/HDV co-infection patients occurs in the presence of cirrhosis[59,60]. In contrast, 10%-20% of HBV-related HCC can occur in the absence of cirrhosis and liver inflammation[61]. Under normal circumstances, the innate and adaptive immune responses are activated during an infection or tissue injury and immune cells are recruited to fight against the pathogen and induce wound healing. Following the elimination of the pathogenviacytolytic and non-cytolytic mechanisms, the damaged tissue is repaired through the wound-healing process[62]. However, the persistence of the inflammatory stimuli (e.g.chronic viral infection) or dysregulation of the immune regulatory mechanisms prevents complete wound-healing and causes non-resolving inflammation that may lead to liver complications resulting in autoimmunity, fibrosis, cirrhosis, metaplasia and/or tumor growth[62].
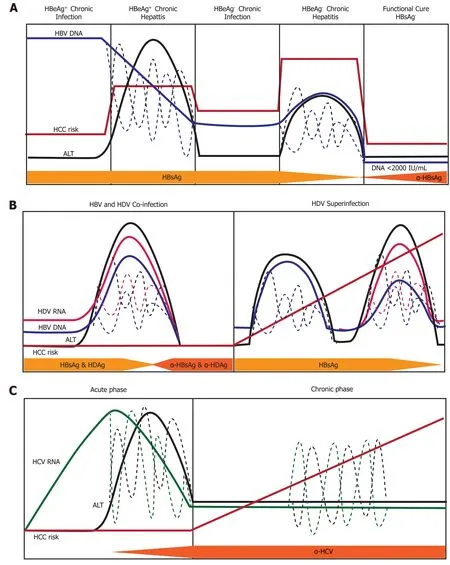
Figure 6 Natural history of infection with hepatitis B, delta, or C virus. Variations in hepatitis B virus (HBV) DNA, hepatitis C virus RNA, hepatitis delta virus (HDV) RNA, and ALT levels indicated by dashed lines. A: Natural history of chronic Hepatitis B virus infection. There are five phases of infection HBeAg+ chronic infection, HBeAg+ chronic hepatitis, HBeAg- chronic infection, and HBeAg- phase. Each clinical phase is defined by a host immune response with respect to HBV viral activity; B: Natural history of HDV infection in either HBV co-infection or HDV superinfection when the individual is a chronic carrier of HBV; and C: Natural history of Hepatitis C virus infection. There are two main phases of infection acute infection and chronic infection. HBV: Hepatitis B virus; HCC: Hepatocellular carcinoma; HDV: Hepatitis delta virus; HCV: Hepatitis C virus.
There are five clinical phases of chronic hepatitis B infection (Figure 6A) from the 2019 AASLD guidelines[63]: HBeAg+chronic infection, HBeAg+chronic hepatitis, HBeAg-chronic infection, HBeAg-chronic hepatitis, and a functional cure (HBsAg-). Each clinical phase is defined by a host immune response with respect to HBV viral activity. During the initial HBeAg+ chronic infection phase the host is immune response has a poorly activated HBV-specific CD8+ T-cell response[64]. Transition to the chronic hepatitis phase is characterized by increased activation of the adaptive immune response (e.g., HBV-specific CD8+T-cells, pro-inflammatory cytokines) which causes decreased HBV DNA levels, liver inflammation, and variable/progressive liver fibrosis. Failure to completely clear HBV in the HBeAg+chronic infection phase results in prolonged over-active immune cell-mediated damage that leads to rapid liver disease progression. Immune-mediated liver damage is facilitated by naturalkiller cells and T-cells through the release of ROS and proinflammatory cytokines which causes bouts of necroinflammation, hepatocyte regeneration/healing and remodeling of the liver microenvironment[65,66]. Constant necroinflammation and failed wound healing responses lead to prolonged oxidative stress exposure which can promote the rapid development of fibrosis, cirrhosis, and cell transformation (epigenetic alterations, oncogenic mutations, telomere shortening, and genomic instability)[67,68]. The 5-year cumulative HCC risk for CHB patients with cirrhosis ranges from 9.7%-15.5%[69]. However, 20% of HCC caused by HBV does not require liver cirrhosis, indicating there are other intrinsic viral associated factors that are responsible for transforming hepatocytes.
HDV infection occurs either in a co-infection model with HBV or as a superinfection from horizontal transmission in individuals with CHB (Figure 6B). The mechanisms used by HDV to modulate the immune system are different from that expressed by HBV and HCV due to the consistent presence of co-infection. The natural history of chronic HDV infection is also dynamic and can be characterized as[70]: (1) Suppressed HBV replication and active HDV replication with high ALT; (2) Slightly decreased HDV replication and HBV reactivation with moderate ALT; and (3) Late-stage disease where cirrhosis and HCC are caused by either HBV/HDV or remission resulting in a reduction of both HBV and HDV viral load. During initial infection, HDV evades IFNα-mediated innate immune responses to promote cell survival and viral persistence[71]. Under normal cellular conditions, double-stranded RNA induces expression IFN-α which binds to the IFN receptor-associated JAK kinase tyrosine kinase-2(tyr-2). Dimerization of the tyr-2 receptor activates a JAK/STAT signaling cascade to produce innate antiviral proteins: myxovirus resistance A, 2’,5’-oligoadenylate synthase, and dsRNA-activated protein kinase[72]. HDV blocks phosphorylation of tyr-2 to prevent downstream signaling and impairs phosphorylation activity and nuclear accumulation of STAT1/STAT2[71].
The superinfection of HDV in patients with CHB has the most severe liver disease outcome, partially due to the pre-existing liver damage caused by HBV infection[73]. Moreover, superinfection with HDV leads to HBV viral load suppression through mechanisms that are not thoroughly understood[74]. Recognition of MHC-1 HDV antigens on infected cells by CD8+T-cells mediates cellular killing. Released viral antigens are endocytosed by Kupffer cells (liver resident macrophages), Blymphocytes, and dendritic cells and presented to CD4+helper T-cellsviaMHC-II receptors. Clonal expansion of CD4+T-cells releases IL-2, IL-10, and IFN-γ cytokines which stimulate immune-mediated killing of HDV infected cells, severe liver necrosis and progressive liver disease[75].
Infection from the HCV is usually acquired from horizontal transmission in adulthood, where 75%-80% of people develop a chronic infection from viral persistence[11]. Chronic hepatitis C (CHC) infected individuals have mild liver inflammation (Figure 6C), stable HCV RNA titers, and liver disease that progresses especially in the presence of other risk factors (age, male, obesity, diabetes alcohol, HIV or HBV co-infection)[76]. HCV infection activates intrinsic type I and III IFN responses which induces transcription of innate-antiviral IFN stimulated genes[77]. Adaptive viral-specific CD8+/CD4+ T-cells and natural killer cells facilitate the release of pro-inflammatory cytokines and growth factors while destroying of HCV infected hepatocytes by promoting the inflammation-necrosis-proliferation cycle[78]. Immunemediated damage produces large amounts of ROS mediated DNA damage, lipid peroxidation, epigenetic modifications, mitochondrial alteration, senescence, and chromosomal translocation that lead to hepatocyte transformation[79]. Immune failure to remove all HCV infected cells causes the selection of viral escape mutants within a carrier population. These escape mutants prevent stimulation of CD4+/CD8+ T-cell responses, and aid in viral immune evasion, chronic infection, loss of immune regulation, and promotion of HCV-mediated HCC[80,81]. Moreover, persistent liver inflammation caused by immune cells over decades of infection can lead to the development of fibrosis, cirrhosis, and HCC. Approximately 10-20% of CHC patients develop cirrhosis in 20-30 years in the absence of treatment for hepatitis C, indicating the high risk for HCC development[82].
Cirrhosis is a major risk factor for HCC development
The liver is made up of approximately 80% parenchymal cells (i.e., hepatocytes) and 20% non-parenchymal cells (e.g., sinusoidal endothelial cells, hepatic stellate cells, and Kupffer cells)[83]. Infection with viral hepatitis primarily targets the large population of hepatocytes, leading to production of ROS. Release of ROS and pro-inflammatory cytokines by Kupffer cells/hepatocytes activate neighboring stellate cells and liver sinusoidal endothelial cells which are key players in the development of fibrosis[84,85](Figure 7). Stellate cells and fibroblasts enhance collagen synthesis and alter the extracellular matrix which lead to remodeling of the liver microenvironment[86].
Progressive inflammation and fibrosis pave the way for disease progression to cirrhosis which is the largest risk factor for HCC development (Figure 7). Cirrhosis is irreversible and often individuals are asymptomatic, which makes diagnosis and treatment difficult[87]. Those who develop severe symptoms of cirrhosis, are likely to have advanced liver disease and HCC. This is especially problematic for the populations who are unaware of their infection status with HBV, HCV, and/or HDV because they are unable to seek treatment intervention to lower the risk of developing cirrhosis. During cirrhosis, altered blood flow can lead to a hypoxic environment for hepatocytes leading to altered molecular signaling and increased oxidative damage[88]. Cells within the context of cirrhosis have experienced a multitude of changes from inflammation mediated damage, repair, and regeneration. The hypoxic environment in the liver during cirrhosis can select for altered oncogenic cells and promote angiogenesis. For a comprehensive review of molecular mechanisms of host factors driving the progression of liver cirrhosis to HCC see (Fridlandet al[88]and Kandaetal[89]).
HBV-SPECIFIC INDUCTION OF HCC
HBV DNA integration
Although HBV uses reverse transcription for replication, unlike retroviruses, integration is not an essential step in the virus lifecycle and does not produce replication-competent virus[90]. During reverse transcription of the pgRNA, partially double-stranded rcDNA is formed 90% of the time[17]. The rcDNA is the genetic material that can be used to replenish the cccDNA pool and produce viable virions that can proceed to infect new hepatocytes. For 10% of cases, the reverse transcription process does not produce rcDNA and instead synthesizes double-stranded linear DNA (dslDNA)[17]. The HBV dslDNA can also be present in virions and repaired to produce cccDNA with a 16nt insertion that cannot produce pgRNA (unless it reverts to wild type cccDNAviahomologous recombination)[91]. Integration of dslDNA is reported to occur in 1 of approximately 105-106infected hepatocytes, and has been observed to occur early in infection (children as young as 5 mo old), and in patients who have acute HBV, CHB, and HCC[23,92]. The currently accepted mechanisms for HBV integration driving HCC include (reviewed by Tuet al[17]): (1)Chromosomal instability from HBV integrated DNA; (2) Insertional mutagenesis in proto-oncogenes and tumor suppressors; and (3) Expression of mutant HBV proteins from integration[17].
A key hallmark of cancer is genome instability. Hepatitis B virus can induce genome instability through viral integration into the host genome to cause cellular transformation (Figure 4). In non-HCC patients, HBV integration sites are randomly distributed through the genome and do not contain enriched sequence mutations. However, in CHB-HCC patients, HBV integration can be enriched in certain areas to cause chromosomal instability through integration near fragile sites: Intergenic regions, repetitive regions (e.g., LINEs), short interspaced nuclear elements, simple repeats, CpG islands, and telomeres[93]. Chromosomal rearrangements and gene copy number variations also contribute to chromosomal instability and are present in the majority of CHB associated HCC[94].
Next-generation sequencing studies that compare HBV integration sites between tumor and matched non-tumor tissues have found that HCC tumors generally have a greater number of integration events and increased integration frequency in coding or promoter regions. In non-tumorous HBV infected hepatocytes, recurrent integration in driver genes can promote hepatocyte clonal expansion[17]. In 10%-15% of HCC cases, recurrent integration of the enhancer II/core HBV promoter in/near telomerase reverse transcriptase (TERT) or myeloid/lymphoid or mixed-lineage leukemia 4 genes causes upregulation of these oncogenes[95,96]. The upregulation of these genes has been observed in early and late tumor development, which may indicate that integration in these genes may aid in cell transformation and HCC progression.

Figure 7 Liver disease progression to hepatocellular carcinoma from chronic viral hepatitis infection. Genetics, co-morbidities, gender, age, and aflatoxin exposure influence liver disease progression along with chronic viral infection with hepatitis B, C, and/or delta virus. Cirrhosis is the greatest risk factor for development of hepatocellular carcinoma, however, hepatocellular carcinoma in the context of chronic Hepatitis B virus infection can occur in the absence of cirrhosis. Chronic hepatitis C infection can lead to steatohepatitis, which can accelerate fibrosis and cirrhosis. Superinfection with Hepatitis delta virus in individuals who have chronic Hepatitis B virus infection creates an accelerated disease course leading to liver failure and/or hepatocellular carcinoma. Many driver mutations (telomerase reverse transcriptase, TP53, CTNNB1, AXIN1, ARID1A/ARID2, NFE2L2/KEAP1/RPS6KA3, KAK1) can occur as liver disease progresses to hepatocellular carcinoma and can lead to accelerated disease progression. TERT: Telomerase reverse transcriptase; HBV: Hepatitis B virus; HDV: Hepatitis delta virus; HCV: Hepatitis C virus.
Integration of HBV dslDNA can lead to the persistent expression of mutant and truncated HBsAg, HBcAg and HBx proteins. High expression rates of these normal and mutated proteins are associated with ER and mitochondrial stress responses which can increase the risk of HCC[97]. These mutants have also been observed to stimulate hepatocyte expansion and may provide a proliferative advantage. In animal models, over-expression of mutant HBsAg and HBx show precancerous liver lesions and HCC[98]. Moreover, expression of C-terminal truncated HBx protein from integrated HBV induces stem-cell-like properties, cell transformation, tumor invasion, and inhibition of apoptosis[17,99].
Deregulation of cellular pathways by HBx protein
The HBx protein (17 kDa) plays various roles in the HBV lifecycle and HCC development (Figure 4)[100]. HBx does not directly bind to DNA, instead, it interacts with other proteins to cause promiscuous transactivation of viral and cellular genes[101]. There are four main mechanisms used by HBx that contribute to HCC[102]: (1) Integration of HBx gene into the hepatocyte genome promoting genetic instability (Figure 4); (2) Interaction with the mitochondrial and other cellular proteins to induce oxidative stress; (3) Activation of cell survival signaling pathways and inactivation of tumor-suppressors; and (4) HBx induced epigenetic modifications such as DNA methylation, histone acetylation, and microRNA expression.
HBx modulates proto-oncogenic signaling pathways that are involved in inflammation and proliferation: Mitogen-activated protein kinase (MAPK)/ Ras/Raf/c-Jun, NF-κβ, JAK-STAT, protein kinase C, Src, survivin and PI3K cascades[101,103]. HBx has also been proposed to activate the Wnt/β-catenin pathway through the binding of Antigen presenting cells protein or inactivation of GSK-3β through Extracellular signal regulated kinase activation. These mechanisms result in the accumulation of β-catenin and increased transcription of pro-angiogenic/metastatic factors[104,105]. HBx promotes genome instability through inhibition of UV-induced DNA damage repair pathways and S-phase progression by binding to UV-DDB1[106]. Hypoxic cirrhotic nodules expressing HBx promotes survival and growth through transcriptional activation/stabilization of HIF1α, which activates transcription of Ang-2 and vascular endothelial growth factor to promote angiogenesis and metastasis[107]. HBx also upregulates matrix metalloproteinases that digest fibrous capsules in tumors resulting in increased epithelial-mesenchymal transition (EMT) and metastasis[108]. HBx can also trans-activate cAMP-response element binding protein response element genes and Yes-associated protein, which are often over-expressed in HCC[109]. The tumor suppressor p53 can also be bound by HBx in the cytoplasm and prevent p53 nuclear localization. Binding of p53 by HBx causes deregulation of cellcycle checkpoints, inhibition of p53 dependent apoptosis and DNA-repair pathways. Loss of p53 activity leads to genome instability and the deregulation of tumor suppressors[47].
HBx is an epigenetic regulator of DNA hyper or hypomethylation in protooncogenes and tumor suppressors, respectively[110]. Viral-induced upregulation of DNMTs causes aberrant hypermethylation of CpG islands in tumor suppressor, leading to gene silencing[111]. In one study, 82% of HCC tumors had at least one tumor suppressor gene inactivated by hypermethylation, indicating the important role of epigenetic modifications in cancer development[112]. HBx protein increases the transcription of methyl catalase DNMT1, which hypermethylates the tumor suppressor gene E-cadherin and INK4A[113]. Loss of INK4A leads to loss of cell-cycle regulation, and loss of E-cadherin promotes epithelial to mesenchymal transition which promotes invasion and metastasis. HBx has also shown to promote histone acetylation and deacetylation to alter the expression of cancer-related genes, microRNAs, and non-coding RNAs. Increased levels of miR-29a, miR143, miR-148a, and miR-602 by HBx promotes upregulation of genes involved in angiogenesis and metastasis[114]. There are several miRNAs that are downregulated by HBx, one of the most important being miR-122 a liver-specific miRNA that has an anti-tumorigenic role[115]. HBx also induces expression of long non-coding RNAs: LINE1 which upregulates Wnt/B-Catenin (promoting invasion and metastasis), HULC, UCA1 which inhibit tumor suppressors p18 and p27 (promoting G1/S cell cycle transition), and DBH-AS1 which activates extracellular signal-regulated kinase (ERK)/ p38/MAPK (anti-apoptosis)[114].
HBV proteins mediate intracellular oxidative stress
Individuals with CHB exhibit 1.5-4 times higher levels of oxidative stress (8-oxoguanine DNA products, lipid peroxidation, oxidation of proteins, decreased levels of anti-oxidant enzyme glutathione and higher oxidative forms) in the liver and plasma/sera compared to HBV negative individuals[116,117]. Extracellular oxidative stress can be immune mediated through the expression of pro-inflammatory cytokines or the release of ROS from cellular destruction. Intrinsic oxidative stress in the ER and mitochondria can be mediated by HBV associated proteins HBsAg, HBcAg, and HBx (Figure 4)[118]. These HBV associated proteins can be expressed from integrated HBV DNA or from the cccDNA minichromosome.
During the HBV lifecycle, secretory proteins such as the HBsAg and HBeAg are folded and assembled in the ER and transported through the Golgi[119]. High expression levels of secretory proteins or mutant HBV proteins that are misfolded can accumulate in the ER and cause activation of an unfolded protein response (UPR)[120]. The UPR induces inflammation, tissue damage from cell death, regeneration, and fibrosis (Figure 7). The wild-type and mutant LHBsAg and mutant HBcAg induce oxidative stress through protein accumulation in the ER membrane causing an UPR[121]. Excess LHBsAg leads to the blockage of HBsAg secretion, while mutant LHBsAg leads to ER stress, which may induce DNA damage and genomic instability[121-123]. A study of a Korean cohort with CHB genotype C suggested mutations in the HBcAg could upregulate ER stress resulting in ROS, increased proinflammatory cytokines, and increased intracellular Ca2+[124]. Activation of the UPR response by HBsAg and HBcAg causes release of hydrogen peroxide and calcium ions into the cytoplasm, enhancing ROS production[121]. HBV infection also reduces antioxidative stress response pathways (e.g.Nrf2/ARE, catalase, and HO-1)[125]. Moreover, HBsAg is able to promote cell transformation through immune dysregulation, upregulation of survival signaling pathways, activation of transcription factors (NFkB, AP-1, STAT3), increased mutations through the generation of free-radicals, cellcycle deregulation, release of pro-inflammatory cytokines, and activation of stellate cells[67,126,127].
The HBx protein can localize into several cellular compartments (mitochondria, cytoplasm, and nucleus) to aid in various roles including transcription, cell-cycle progression, protein-degradation, apoptosis, and genetic instability[100]. Localization of HBx on the outer mitochondrial membrane, causes reduced expression and activity of respiratory complex proteins I, II, IV and V in the electron transport. Reduced cellular respiration results in altered mitochondrial function, increased production of superoxide anions, and 8-oxoguanine DNA products[118,128,129].
HDV-SPECIFIC INDUCTION OF HCC
HDV can indirectly mediate hepatocarcinogenesis through innate immune response modifications, induction of adaptive immune responses, epigenetic changes, lncRNA modifications and ROS production (Figure 4). The L-HDAg has an important role in facilitating many of these mechanisms through interaction with signaling pathways involved in pro-growth/survival, apoptosis, and wound healing[130,131]. Activation of the transforming growth factor β (TGF-β) and AP-1 pathways by L-HDAg binding of Smad3, STAT3, and c-jun promotes EMT, fibrosis, and cell-transformation[131,132].
HDV is also able to promote oxidative stress in the ER through L-HDAg’s interaction with NOX-4[133]. Activation of the NOX4 pathway causes the release of ROS which can activate STAT3 and NF-κβ signaling[133]. The L-HDAg can also promote proinflammatory NF-κβ activity through stimulation of TNF-α. The S-HDAg can directly bind to glutathione S-transferase P1 mRNA causing downregulation in expression, increased ROS, and apoptosis[134]. Moreover, epigenetic modifications such as histone H3 acetylation by small and large HDAg enhances clusterin gene expression[128]. Increased levels of clusterin and histone acetylation aid in HDV infected cell survival and are upregulated in cancerous cells[135,136].
HCV-SPECIFIC INDUCTION OF HCC
Viral protein-mediated oxidative stress
Similar to HBV, individuals with chronic HCV infection experience significant decreases in antioxidant enzymes, and two to seven logs increase in liver and blood oxidative stress[137,138]. Prolonged oxidative stress results in increased levels of free oxygen radicals, DNA adduct formation (e.g.8-oxoG), protein adducts, and lipid peroxidation[116,139,140].
In the ER, oxidative stress is mediated by HCV core, E1, E2, NS4B, and NS5A proteins (Figure 5). Viral glycoproteins E1/E2, and non-structural protein NS4B induce the unfolded protein response in the ER, which causes calcium release and production of hydrogen peroxide[141,142]. NS5A facilitates calcium uptake in the mitochondria and ER, causing release of hydrogen peroxide and organelle dysfunction[143]. HCV core-mediated binding of the mitochondria activates the mitochondrial calcium uniporter facilitating the uptake of ER released calcium ions[144]. Subsequently, an influx of calcium into the mitochondria directly effects the electron transport chain and leads to increased ROS production[144,145].
Enhanced expression of TGF-β1 from HCV core and NS5A upregulates the production of Nicotinamide adenine dinucleotide phosphate oxidases NOX1/NOX4 and cytochrome p450 2E1 oxidase (CYP2E1)[146,147]. CYP2E1 aids in metabolism of ethanol and drugs with the release of superoxide and hydrogen peroxide byproducts[148]. Although CYP2E1 has an important metabolic role, high levels of expression induced by HCV core and NS5A lead to increased levels of ROS byproduct accumulation[146,149]. In the context of HCV induced fibrosis, CYP2E1 expression levels are also increased which may imply that ROS have an important role in liver damage[150]. NOX1 expression and localization to the nuclear membrane is stimulated shortly after HCV infection and promotes release of superoxide ions into the cytoplasm[151]. NOX4 can be found on the nuclear or the ER membranes, which release hydrogen peroxide into the cytoplasm or nucleus promoting direct DNA damage[151].
Hepatic steatosis
Steatohepatitis is characterized by the presence of excess triglycerides in hepatocytes. HCV promotes steatohepatitis through enhancing lipogenesis, and impairing lipid degradation/export which may cause cellular lipotoxicity[152]. Approximately 40%-80% of CHC individuals have steatohepatitis due to viral pathogenesis, which is associated with the increased risk of HCC[153]. The HCV core protein is a key player in altering lipid metabolism through (Figure 5): (1) Decreasing lipid turnover of HCV core particles coated lipid droplets (LD); (2) Inhibition of LD mobility; (3) Inhibition of microsomal triglyceride transfer protein which prevents lipid export and degradation; and (4) Inhibition of peroxisome proliferator-activating receptor-α/γ; inhibition of diacylglycerol acetyltransferase 1[154,155]. Accumulation of free fatty acids causes severe mitochondrial and ER oxidative stress. The accumulation or ROS stimulates lipid peroxidation and activation of inflammatory signaling cascades such as TNF-α and IL-1 which can lead to the development of steatohepatitis and insulin resistance[156].
Deregulation of cellular pathways by HCV proteins
Activation of cell-survival and growth pathways are mediated by core, E2, NS2, NS3, NS4A NS5a, and NS5B proteins (Figure 5). To promote cell cycling and evasion of the G1/S checkpoint, NS5B binds to tumor suppressor Rb to facilitate proteasomal degradation and release of E2F to produce cell-cycle dependent genes[157]. NS2 activates the cyclin D/CDK 4 complex to induce the expression of cyclin E[158]. The core protein upregulates cyclin E/CDK 2 to promote cell cycle transition from G1 to S phase with checkpoint evasion, genome instability, and aberrant cell growth[159]. NS5A inactivates the tumor suppressor pTEN through binding, to cause proliferative growth and survival using the PI3K/Akt pathway[160]. HCV Core, E2, NS3, and NS5A interact with various proteins in the RAF/MAPK/ERK pathways to promote cellular proliferation[161-164]. The Wnt/β-catenin signaling pathway is activated by direct binding of β-catenin by NS5A or phospho-inactivation of GSK-3β by NS5A and core proteins[165-168]. Activation of Wnt target genes promotes proliferation, angiogenesis and EMT transfer. High quantities of β-catenin are associated with poor prognosis of HCC[169].
The inhibition of apoptosis contributes to the development of HCC through the growth of abnormal cells. The tumor suppressor protein, p53, is targeted by many HCV proteins to prevent apoptosis, DNA-repair, and senescence. NS5A directly binds to p53 causing inhibition, while NS2, NS3/4A interfere with the p53 pathway by inducing p53 delocalization from the nucleus to the cytoplasm or perinuclear regions[170-173]. There is some evidence that the HCV core protein is also able to bind to p53, however, this is debated, because high levels of core cause repression while low levels promote p53 activity[174]. To avoid cell death, HCV also has various protein mechanisms to inhibit TNF-α cytokine-mediated apoptosis: (1) The core protein activates FLICE, an inhibitor of TNF-α signaling[175]; (2) NS5A protein prevents TNF-αmediated cell death by inhibiting activation of caspase-3 and PARP1 cleavage[176]; and (3) NS5A can also interact with intrinsic apoptosis regulator Bid to inhibit activation of apoptosis[177].
The expression of TGF- β signaling is antiproliferative and pro-apoptosis[178]. HCV NS5A binds directly to the TGF- β receptor 1 to block signaling, and as such prevents phosphorylation and nuclear localization of smad2 and the smad3/smad4 heterodimer[179]. Mutant core proteins derived from HCV tumors inhibit the TGF- β pathway through direct interaction with Smad3, which results in the inhibition of DNA-binding by the Smad3/4 heterodimer[180]. Inhibition of the TGF- β pathway promotes EMT which enhances fibrogenesis, tumor invasion, and metastasis[178].
COMMON SOMATIC MUTATIONS IN PROGRESSIVE HCC TUMORS
As the progression of liver disease to HCC occurs, many common driver mutations that allow for selective growth advantage for tumor cells over normal cells can be identified (Figure 7). HCC tumors are highly heterogeneous within the same individual and these differences in tumor genetic profiles are amplified by single cell and next-generation sequencing. From sequence analysis several driver genes have been identified in the progression of HCC: TERT, tumor protein p53 (TP53), catenin beta 1 (CTNNB1), Wnt/β-catenin signaling protein AXIN1, chromatin remodeling genes ARID1A and ARID2, oxidative stress response genes NFE2L2 and KEAP1, RAS/MAPK signaling (RPS6KA3), and the JAK/STAT signaling cascade activator (KAK1). The most disrupted driver genes are described below, for a comprehensive overview refer to[181].
Regardless of geographic location, recurrent somatic mutations in the TERT promoter have been identified as the most common mutation in HCC (20.7%-59%)[181]. The TERT protein has an important role in maintaining telomere length by adding short-repeated TTAGGG nucleotides at the end of chromosomes[182]. Maintenance of the telomeres is important to avoid DNA damage, however normal adult cells do not express TERT and can only undergo 40-60 cycles of replication before senescence[183]. In HCC, activating mutations in the TERT promoter enable replicative immortality through the consistent addition of telomeric repeats which allow cells expressing TERT to replicate without entering senescence[182,183]. TERT mutations have been identified to occur early in malignant transformation and persist throughout tumor progression[184]
The tumor suppressor protein P53 is a critical protein commonly mutated in cancer, that is involved in cell-cycle arrest at the G1/S checkpoint and activation of apoptosis[185]. TP53 is most frequently mutated in its DNA binding domain, to prevent its role in activating TP53 responsive genes that aid in cell-cycle control[186]. Loss of p53 function aids cell transformation through constant cell cycling without DNA damage checkpoint regulation[5]. Mutational frequency of P53 in HCC is dependent on geographic locations, as mutational frequency requires aflatoxin exposure. High dietary aflatoxin exposure and endemic hepatitis B infection is associated with TP53 mutations located on R249S/V157F and poor prognosis[187,188].
High mutational frequency in the CTNNB1 gene which codes for the β-catenin transcription factor in the Wnt-pathway is associated with tumor progression and poor prognosis[5]. Activating mutations in CTNNB1 increase cytoplasmic accumulation of βcatenin without a Wnt signaling molecule[189]. Normally without a signaling molecule, β-catenin is expressed, bound by the destruction complex, phosphorylated by GSK3, and degraded by the proteasome[189]. However, in the presence of a Wnt ligand signal, β-catenin is not degraded and instead it is expressed to high levels and translocates into the nucleus and activates transcription factor TCF to transcribe Wnt target genes[190]. These target genes are involved in growth, proliferation, and EMT promoting metastasis[5,191]. In HCC, the tumor suppressor AXIN1 is the second most mutated gene (6.8%) in the Wnt-signaling pathway. AXIN1 is involved as a scaffold protein for the β-catenin destruction complex, and inactivating mutations prevent the complex from forming and destroying β-catenin, thus leading to increased β-catenin levels leading to tumor growth and proliferation[190,192]. The deregulation of Wnt/βcatenin signaling has been observed in 40%-70% of HCC patients. Moreover, β-catenin associated mutations occur in lower frequencies in HBV-related HCC, and are more common in HCV and alcohol-related HCC[181].
The ARID1A and ARID2 genes encode for a key subunit component of the SWI/SNF chromatin remodeling complex[193]. The dysregulation of the SWI/SNF complex contributes to tumor heterogeneity, and drug treatment resistance[181]. ARID1A acts as a tumor suppressor gene that is expressed at high levels in normal livers to regulate DNA activity using nucleosomes to restrict cell proliferation[5]. Inactivating mutations in the ARID1A and ARID2 genes occur in around 10% of HCC cases and are associated with poor prognosis, increased cell proliferation, and migration/metastases of HCC cells[181,193,194].
CONSIDERATIONS FOR THE FUTURE
The low 5-year survival rate for those diagnosed with HCC is largely due to the failure of early detection of small lesions and lack of medical therapy for advanced disease[195]. The best curative treatment option for HCC is liver transplantation, however, this is limited to those who are in the early stages of HCC and follow the Milan transplantation criteria or the Alberta HCC algorithm in Canada[196,197]. Other available treatment options during earlier stages of HCC include surgical resection (70% 5-year survival rate) and radiofrequency ablation therapy (40%-70% 5-year survival rate in tumors < 2 cm)[198,199]. During intermediate HCC disease stages, transarterial chemoembolization treatment can be offered (median survival rate 16-20 mo)[200]. In advanced disease, HCC is often quite unresponsive to most chemotherapeutics, and current chemotherapeutics (Sorafenib and Lenvatinib) that target overexpressed receptor-tyrosine-kinase pathways (e.g. vascular endothelial growth factor, MAPK, EGFR, RAS, IGF, PI3K/PTEN/Akt/mTOR, Wnt/beta-catenin) only increase median survival by three months[201]. In 2018, the monoclonal antibody nivolumab which targets the programed cell death 1 receptor on T-cells was approved as a second-line therapeutic for HCC. Nivolumab has a 20% response rate in initial phase II clinical trials and works by activating T-cells for the immune-mediated killing of tumors[196].
Exploiting traits of virally induced cancers as therapeutics
An important feat for the future of HCC treatment will be the development of effective immunotherapies. Nivolumab was the first monoclonal antibody approved for advanced-stage HCC treatment and there are many others that are currently in clinical trials. Since both chronic viral infection and cancer create an immunosuppressive environment to prevent cytotoxic killing, future immunotherapies could target regulatory (Treg) and resident memory T-cells (TRM) to reactivate the immune system against viral infection and HCC tumors[202]. Additionally, molecular pathways deregulated by HBV/HCV/HDV could be targeted by immunotherapies through inhibition of pathways involved in aberrant cell growth leading to HCC. Alternatively, tumors caused by viral etiologies can be targeted through training of the immune system to target viral particles and/or fusion proteins in HCC[203]. Tanet al[203], describes a CAR-T cell technology that can recognize HBV specific epitopes in HCC tumors. Since HBV-associated tumors do not contain actively replicating viruses and only express partially integrated/truncated proteins, T-cells can be designed to target these tumors associated antigens. One of the patients from this study treated with HBV specific CAR-T cells had a decreased tumor volume in 5 of 6 pulmonary metastases over the course of 1-year. Building on the study performed by Tanet al[203], another possibility to improve the persistence of CAR-T technology in viral related HCC could be through engineering a separate CAR-T receptor to recognize viral antigens to boost T-cell populations while targeting cancer-specific lesions. Although immunotherapeutics for solid tumors is in its infancy, this is likely the future for the development of better treatment options for HCC.
CONCLUSION
Chronic hepatitis B, C, and delta viral infections affect almost half a billion people worldwide. Decades-long persistent viral infection and immune-mediated damage cause significant changes in the liver microenvironment and are the strongest risk factors for the development of HCC. Current treatment options for HBV and HCV reduce HCC risk, but do not eliminate it. Moreover, the lack of an HBV virological cure and limited treatment options for HDV requires the exploration of more effective treatments.
HBV and HCV can manipulate pathways in ten hallmarks of cancer, which may explain how these viruses escalate risk of HCC development. HDV is not considered a “directly” oncogenic virus, due to HDV reliance on HBV to complete the viral life cycle, uses several mechanisms to aid the progression of liver disease and increase the risk of HCC[46]. Successful epidemiology studies,in-vitrocell culture studies, and animal studies have provided us with significant insight into the molecular mechanisms of interactions between host-viral interactions. Genomic analyses comparing HCC tumors to those of healthy tissue have provided us insight into driver mutations that aid in the progression of viral-mediated HCC and possible targets for future treatments. Although much progress has been made in the field, there is a lot that remains unknown due to the lack of cell-culture systems that can be used to study all viral genotypes, co-infections, and animal models that can be infected with these viruses to produce liver disease similar to humans. The development of stronger experimental models will provide us with further insight into the role these viruses play in promoting HCC development. Until we have better screening methods and more accessible and/or effective antiviral treatments, the rates of liver cancer will be steadily on the rise. Thus, we need to investigate commonly deregulated pathways in HCC to identify targets and develop more effective treatments to improve the survival rate for those diagnosed with this disease.
ACKNOWLEDGEMENTS
Viral life cycle figures were produced using BioRender Premium.
杂志排行

World Journal of Gastroenterology的其它文章
- Role of betaine in liver disease-worth revisiting or has the die been cast?
- Management of an endoscopy center during the outbreak of COVID-19: Experience from West China Hospital
- Gastrointestinal complications after kidney transplantation
- Is vitamin D receptor a druggable target for non-alcoholic steatohepatitis?
- Acetyl-11-keto-β-boswellic acid inhibits proliferation and induces apoptosis of gastric cancer cells through the phosphatase and tensin homolog /Akt/ cyclooxygenase-2 signaling pathway
- Endogenous motion of liver correlates to the severity of portal hypertension