甲萘胺衍生物的C-8选择性氧甲酰甲基化反应
2020-11-18戚天木吴依彤
程 凯 戚天木 吴依彤
(绍兴文理学院 精细化学品传统工艺替代技术研究重点实验室,浙江 绍兴 312000)
0 引言
甲萘胺类化合物是一类重要芳香胺化合物,是染料合成中的重要中间体[1],也是合成农药中间体和多种橡胶防老剂的重要原料[2].甲萘胺类衍生物在有机合成中也是重要的催化剂和合成原料,在许多农药和功能材料中有诸多应用[3].因此,对甲萘胺类衍生物结构上的修饰及拓展是非常重要的.
近几年来,对于合成甲萘胺类衍生物的方法有诸多报道,主要是在过渡金属催化下,通过碳氢活化的方法,对甲萘胺的C-2[4]、C-4[5]和C-8位[6]上进行修饰而进行合成.由于经典有机化学中甲萘胺的亲电取代反应在α位,所以反应位点选择性较差,主要生成C-4取代产物,并混合有C-6和C-8取代产物.而通过双齿导向基团辅助的钯催化C-H活化反应,可以实现对甲萘胺C-8位进行修饰而实现选择性官能团化[7].(图1)双齿导向基团通过金属络合作用能够稳定过渡金属的高氧化态,从而促进C-H键官能化,而且导向基团在反应结束后也容易去除,是一种简单高效、高选择性的合成C-8取代的甲萘胺衍生物的方法[8].2013年,齐陈泽课题组[9]报道了在N,N-双齿螯合导向下和二价钯的催化下,选择性活化了甲萘胺萘环上C-8位的C-H键,以碘代芳烃作为芳基化试剂,以2-吡啶甲酰-1-萘胺或2-喹啉甲酰-1-萘胺作为底物,实现了甲萘胺上C-8位的选择性芳基化反应.次年,该课题组[10]又报道了2-吡啶甲酰-1-萘胺或2-喹啉甲酰-1-萘胺作为底物,以碘代烷烃为烷基化试剂,实现了甲萘胺上C-8位的选择性烷基化反应.上述报道中实现了1-萘胺衍生物在双齿导向基和二价钯的作用下,实现了C-8位的选择性芳基化和烷基化,但都是以碘代芳烃和碘代烷烃进行反应,而对于价廉易得的氯代烷烃则并不适用.与C-I键相比,C-Cl键更难于跟过渡金属发生氧化加成而参与反应,所以让氯代烷烃参与C-H官能化反应,是十分具有挑战性的.本文首次报道了在二价钯的催化下,2-喹啉甲酰-1-萘胺与氯乙酸甲酯的反应,实现C-8位氧甲酰甲基取代甲萘胺衍生物的合成,提供了一种甲萘胺衍生物C-8位烷基化的新途径.
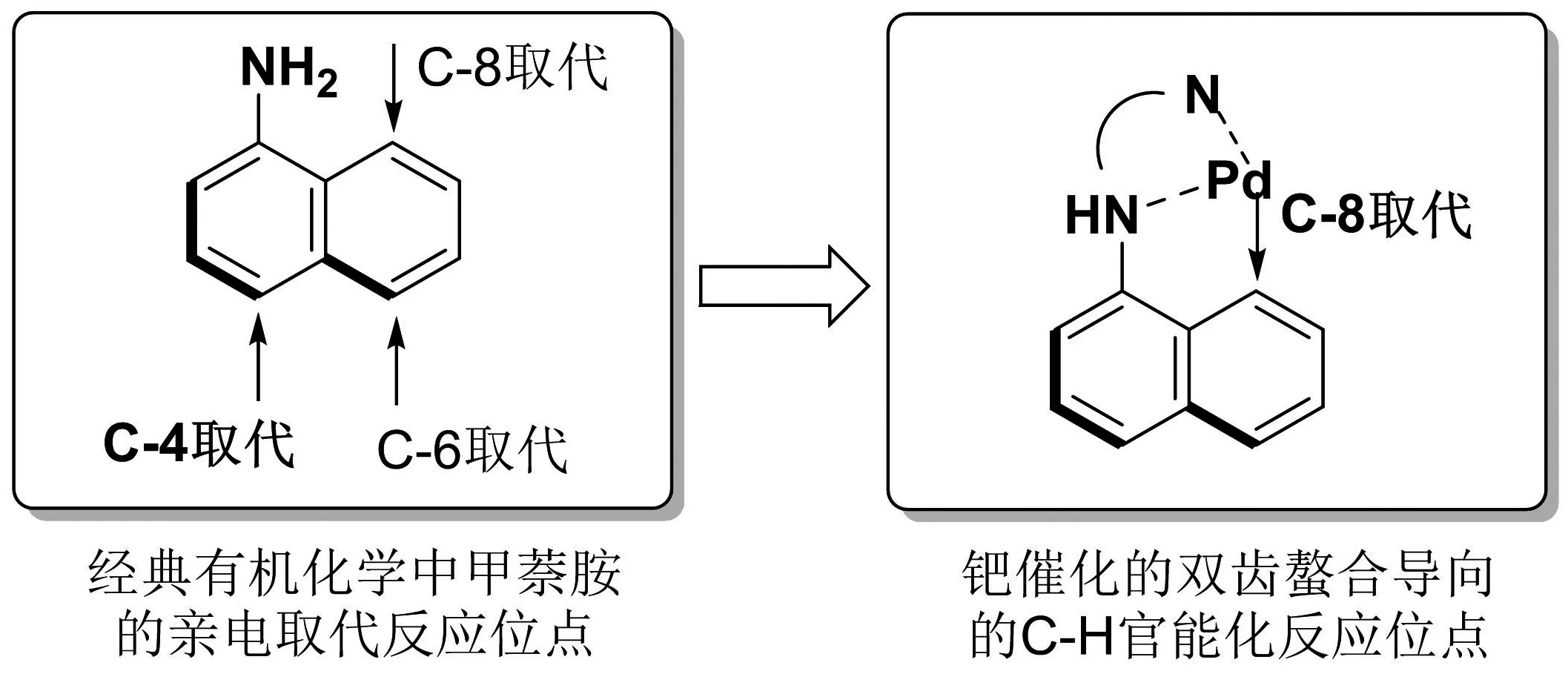
图1 钯催化的双齿螯合导向实现甲萘胺的选择性C-8位官能化
1 实验部分
1.1 试剂与仪器
所有试剂均从试剂公司购得,均为分析纯,未经任何纯化处理直接使用.薄层硅胶板采用GF254(青岛海洋化工厂),用UV(波长254 nm)显色,快速柱层析用烟台江友硅胶(300-400).一般使用乙酸乙酯和石油醚作为展开剂,操作中采用的比例为两者的体积比.1H NMR采用核磁共振仪(Bruker Advance DMX400(400 MHz)型)测定,内标为TMS,溶剂采用氘代氯仿(CDCl3);气相色谱-质谱联用仪采用美国安捷伦(6890N/5975MSD).
1.2 实验过程
1.2.1 原料2-喹啉甲酰-1-萘胺的合成
将喹啉-2-羧酸、1-萘胺、Et3N以摩尔比为1∶1∶2的比例溶于CH2Cl2中,在冰水浴的条件下滴加POCl3,搅拌反应0.5 h后,移至室温下反应约2 h,使其反应完全.反应结束时,将反应混合物冷却至0 ℃,缓慢加入冰水淬灭反应.反应产物在有机相,通过CH2Cl2萃取水相分离收集有机相,用饱和NaHCO3洗涤和MgSO4干燥.最后通过减压蒸发溶剂CH2Cl2后,通过冷却结晶得到产物2-喹啉甲酰-1-萘胺.
1.2.2 反应条件优化
初步尝试选取2-喹啉甲酰-1-萘胺1 a与氯乙酸甲酯2 a作为标准反应底物,在10 mol%的醋酸钯作为催化剂,苯甲酸钾为碱,二氯乙烷作为反应溶剂的条件下,经90 ℃反应12 h.通过薄层色谱显色发现有二种新物质生成,经柱层析色谱分离和核磁共振仪鉴定后,发现目标产物有39%的收率,且有副产物生成.考察其他溶剂,发现叔丁醇作为溶剂的条件下,目标产物的收率为59%,且没有副产物产生,因此我们对醇类反应溶剂进一步筛选,结果如表1所示,以叔丁醇为反应溶剂的时候,产物收率最高,异丙醇的产率稍低,而其他考察的醇类不适用于该反应.因此确定叔丁醇为最优反应溶剂.
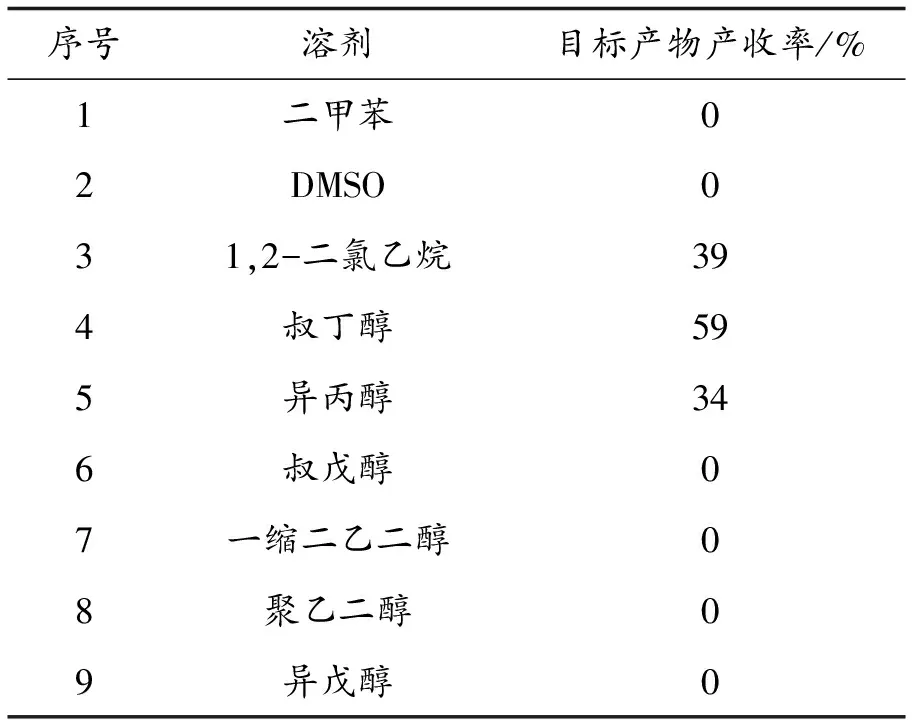
表1 醇类反应溶剂的筛选
接着,对反应的催化剂进行筛选,结果如表2所示,发现在Pd(OAc)2为催化剂的条件下,目标产物收率最高,Pd(dppf)Cl2催化下有45%的产率,Pd(MeCN)Cl2催化下反应产率下降至23%.因此确定了Pd(OAc)2为最优反应催化剂.当催化剂的量下降至5mol%时,反应产率由59%下降至42%.

表2 反应催化剂的筛选
对反应的碱筛选结果如表3所示,发现碱为苯甲酸钾的条件下,目标产物收率最高,而在醋酸钾存在下产率较低,而其他碱对反应无效果.因此确定了苯甲酸钾为最优反应碱.

表3 反应碱的筛选
最后,对反应温度进行了筛选,筛选结果如表4所示,固定其他反应条件,反应温度为90 ℃时,目标产物收率最高,因此确定了90 ℃为最优反应温度.
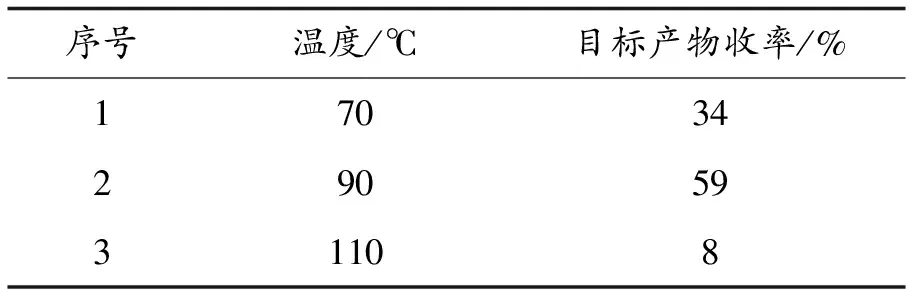
表4 反应温度的筛选
得到了最优反应条件,反应催化剂为醋酸钯,反应碱为苯甲酸钾,反应溶剂为叔丁醇,反应温度为90 ℃,如图2所示.将0.1 mmol,1.0 equiv.2-喹啉甲酰-1-萘胺,0.2 mmol,2.0 equiv.氯乙酸甲酯,0.01 mmol,0.1 equiv.醋酸钯和0.2 mmol,2.0 equiv.苯甲酸钾置于干燥的反应试管中,注入2 mL叔丁醇作为溶剂,密封后将反应管置于90 ℃油浴中加热12 h,反应结束后使体系冷却至室温,用二氯甲烷润洗抽滤柱分离得到目标产物.

图2 反应示意图
1.2.3 反应底物拓展
在最优条件的基础上,对反应底物的广泛性进行了探究.改变酯基上取代基团的结构,对反应活性的影响不明显.无论甲基,乙基,苄基还是大位阻的异丙基和叔丁基,都能顺利地发生预期反应.当氯乙酸甲酯的α位引入甲基取代基,产率明显降低,说明α位的空间位阻对二价钯与氯乙酸甲酯的氧化加成较为困难,降低了反应效率,如表5所示.

表5 反应底物拓展
1.3 产物表征数据
Methyl 2-(8-quoinoline-2-carboxamido)naphthalen-1-yl)acetate:1H NMR(400 MHz, CDCl3)δ 10.81(s, 1 H), 8.43(d,J=8.0, 1 H), 8.37(d,J=8.4 Hz,1 H), 8.14(d,J=8.4 Hz, 1 H), 7.91(d,J=7.6 Hz,1 H), 7.81(m, 4 H), 7.64(t,J=7.2 Hz, 1 H), 7.55(t,J=7.2 Hz, 1 H), 7.40(t,J=7.2 Hz,1 H),7.33(d,J=6.4 Hz, 1 H), 4.35(s, 2 H), 3.62(s, 3 H);13C NMR(100 MHz, CDCl3)δ 173.1, 164.1, 150.2, 146.6, 137.7, 136.2, 132.4, 131.3, 130.3, 129.7, 129.6, 129.5, 129.1, 129.0, 128.7, 128.2, 127.9, 127.1, 125.6, 125.3, 119.3, 52.3, 41.8.HRMS(ESI)Calcd for C23H18N2O3[M + H]+371.1390, found 371.1392.
Ethyl 2-(8-quoinoline-2-carboxamido)naphthalen-1-yl)acetate:1H NMR(400 MHz, CDCl3)δ 10.87(s, 1 H), 8.44(d,J= 8.0, 1 H), 8.39(d,J= 8.0 Hz, 1 H), 8.15(d,J= 8.0 Hz, 1 H), 7.93(d,J= 8.0 Hz, 1 H), 7.83(m, 4 H), 7.66(t,J= 6.8 Hz, 1 H), 7.55(t,J= 7.4 Hz, 1 H), 7.41(t,J= 7.2 Hz, 1 H), 7.34(d,J= 6.4 Hz, 1 H), 4.34(s, 2 H), 4.11(q,J= 6.8 Hz, 2 H), 0.97(t,J= 6.4 Hz, 3 H);13C NMR(100 MHz, CDCl3)δ 172.8, 164.1, 150.2, 146.6, 137.7, 136.2, 132.5, 131.2, 130.3, 129.7, 129.5, 129.4, 129.2, 129.0, 128.6, 128.2, 127.9, 127.0, 125.5, 125.3, 119.3, 61.1, 42.0, 13.8.HRMS(ESI)Calcd for C24H20N2O3[M + H]+385.1547, found 385.1549.
Isopropyl 2-(8-quoinoline-2-carboxamido)naphthalen-1-yl)acetate:1H NMR(400 MHz, CDCl3)δ 10.81(s, 1 H), 8.43(d,J= 8.0, 1 H), 8.37(d,J= 8.4 Hz, 1 H), 8.14(d,J= 8.4 Hz, 1 H), 7.91(d,J= 7.6 Hz, 1 H), 7.81(m, 4 H), 7.64(t,J= 7.2 Hz, 1 H), 7.55(t,J= 7.2 Hz, 1 H), 7.40(t,J= 7.2 Hz, 1 H), 7.33(d,J= 6.4 Hz, 1 H), 4.35(s, 2 H), 3.69(m, 1 H), 1.28(d,J= 7.2 Hz, 6 H);13C NMR(100 MHz, CDCl3)δ 173.1, 164.1, 150.2, 146.6, 137.7, 136.2, 132.4, 131.3, 130.3, 129.7, 129.6, 129.5, 129.1, 129.0, 128.7, 128.2, 127.9, 127.1, 125.6, 125.3, 119.3, 52.3, 41.6, 27.6.HRMS(ESI)Calcd for C25H22N2O3[M + H]+398.1630, found 398.1635.
Tert-butyl 2-(8-(quoinoline-2-carboxamide)naphthalen-1-yl)acetate:1H NMR(400 MHz, CDCl3)δ 11.11(s, 1 H), 8.42(d,J= 8.4 Hz, 1 H), 8.35(d,J= 8.4 Hz, 1 H), 8.19(d,J= 8.8 Hz, 1 H), 7.90(t,J= 8.0 Hz, 2 H), 7.79(m, 3 H), 7.62(t,J= 7.4 Hz, 1 H), 7.53(t,J= 7.6 Hz, 1 H), 7.37(m, 2 H), 4.35(s, 2 H), 1.29(s, 9 H);13C NMR(100 MHz, CDCl3)δ 171.8, 164.0, 150.4, 146.6, 137.7, 136.2, 132.6, 131.0, 130.1, 130.0, 129.8, 129.4, 128.5, 128.4, 128.1, 127.8, 126.5, 125.5, 125.3, 119.4, 81.5, 43.3, 28.0.HRMS(ESI)Calcd for C26H24N2O3[M + H]+413.1860, found 413.1864.
Benzyl 2-(8-(quoinoline-2-carboxamide)naphthalen-1-yl)acetate:1H NMR(400 MHz, CDCl3)δ 10.86(s, 1 H), 8.41(d,J= 8.0 Hz, 1 H), 8.34(d,J= 8.4 Hz, 1 H), 8.06(d,J= 8.4 Hz, 1 H), 7.88(m, 3 H), 7.81(d,J= 7.2 Hz, 1 H), 7.73(t,J= 7.2 Hz, 1 H), 7.64(t,J= 7.2 Hz, 1 H), 7.56(t,J= 8.0 Hz, 1 H), 7.41(t,J= 7.2 Hz, 1 H), 7.35(d,J= 6.8 Hz, 1 H), 7.17(m, 3 H), 6.98(d,J= 6.8 Hz, 2 H), 5.06(s, 2 H), 4.41(s, 2 H);13C NMR(100 MHz, CDCl3)δ 172.6, 164.1, 150.1, 146.6, 137.7, 136.2, 135.3, 132.4, 131.4, 130.3, 129.8, 129.6, 129.4, 129.1, 129.0, 128.7, 128.3, 128.1, 128.0, 127.8, 127.7, 127.1, 125.6, 125.3, 119.2, 66.8, 41.9.HRMS(ESI)Calcd for C29H22N2O3[M + H]+447.1703, found 447.1690.
Methyl 2-dimethyl-(8-quoinoline-2-carboxamido)naphthalen-1-yl)acetate:1H NMR(400 MHz, CDCl3)δ 10.87(s, 1 H), 8.44(d,J= 8.0, 1 H), 8.39(d,J= 8.0 Hz, 1 H), 8.15(d,J= 8.0 Hz, 1 H), 7.93(d,J= 8.0 Hz, 1 H), 7.83(m, 4 H), 7.66(t,J= 6.8 Hz, 1 H), 7.55(t,J= 7.4 Hz, 1 H), 7.41(t,J= 7.2 Hz, 1 H), 7.34(d,J= 6.4 Hz, 1 H), 3.61(s, 3 H), 2.29(s, 6 H);13C NMR(100 MHz, CDCl3)δ 172.8, 164.1, 150.2, 146.6, 137.7, 136.2, 132.5, 131.2, 130.3, 129.7, 129.5, 129.4, 129.2, 129.0, 128.6, 128.2, 127.9, 127.0, 125.5, 125.3, 119.3, 61.1, 42.0, 26.1, 18.3.HRMS(ESI)Calcd for C25H22N2O3[M + H]+398.1630, found 398.1622.
Ethyl 2-dimethyl-(8-(quoinoline-2-carboxamide)naphthalen-1-yl)acetate:1H NMR(400 MHz, CDCl3)δ 11.11(s, 1 H), 8.42(d,J= 8.4 Hz, 1 H), 8.35(d,J= 8.4 Hz, 1 H), 8.19(d,J= 8.8 Hz, 1 H), 7.90(t,J= 8.0 Hz, 2 H), 7.79(m, 3 H), 7.62(t,J= 7.4 Hz, 1 H), 7.53(t,J= 7.6 Hz, 1 H), 7.37(m, 2 H), 4.11(q,J= 6.8 Hz, 2 H), 2.21(s, 6 H), 0.97(t,J= 6.4 Hz, 3 H);13C NMR(100 MHz, CDCl3)δ 171.8, 164.0, 150.4, 146.6, 137.7, 136.2, 132.6, 131.0, 130.1, 130.0, 129.8, 129.4, 128.5, 128.4, 128.1, 127.8, 126.5, 125.5, 125.3, 119.4, 81.5, 43.3, 28.0, 17.4.HRMS(ESI)Calcd for C26H24N2O3[M + H]+413.1860, found 413.1868.
2 反应机理
如图3所示,该反应首先是二价钯和2-喹啉甲酰-1-萘胺(1 a)发生双齿螯合作用,生成中间体A.通过酰胺的氮原子和喹啉的氮原子络合钯,中间体A选择性发生C-H活化,甲萘胺C-8位上的C-H键插入之后形成中间体B,在碱的辅助下与氯乙酸甲酯(2 a)氧化加成形成中间体C,经还原消除之后得到3 b产物.

图3 反应的可能机理
3 结论
综上所述,本文提供了一种以氯代物为烷基化试剂,利用双齿导向基团,选择性地实现了甲萘胺C-8位的烷基化的新方法.通过对2-喹啉甲酰-1-萘胺与氯乙酸甲酯的反应条件优化进行的一系列筛选,确定了最优反应条件,反应催化剂为醋酸钯,反应碱为苯甲酸钾,反应溶剂为叔丁醇,反应温度为90 ℃.在最优反应条件下,可以以59%的较高收率,实现甲萘胺上C-8位的烷基化反应.同时对各种氯乙酸酯类化合物进行底物拓展,发现甲萘胺C-8位的氧甲酰甲基取代都能顺利发生.该合成方法虽然仍具有一定局限性,但是对通过简单易得的氯代物实现的钯催化的双齿螯合导向的甲萘胺衍生物C-8烷基化反应的研究具有较大的参考价值.
