铱催化的MBH 乙酸酯与吲哚酮类化合物的烯丙基化反应
2020-10-16陈相孟张雅琪欧阳嘉盛和晓波普晓云潘本都邱立勤
陈相孟,张雅琪,梁 豪,陈 彬,欧阳嘉盛,和晓波,钱 旭,普晓云,潘本都,邱立勤
(中山大学化学学院,广东省手性分子与药物发现重点实验室,广州510275)
3,3-二取代的氧化吲哚酮衍生物是非常重要的母核结构,存在于很多天然产物、生物碱及药物活性分子的结构单元中.如Mollenine,Physostigmine和Flustramine B等都在吲哚酮的C3位上存在1个线性烯丙基链.如何直接、简便地构建该季碳中心,一直是有机化学科研工作者感兴趣的课题.Morita-Baylis-Hillman(MBH)反应是形成碳-碳键和碳-杂键的基本反应之一.α,β不饱和化合物与醛类化合物的MBH反应产物可作为合成其它有机目标分子的原料,其中芳香醛衍生的MBH乙酸酯在烯丙基取代反应中是比较重要的底物,其酯基比较容易离去,易与不同的亲核试剂发生烯丙位取代反应,且反应条件温和,原子经济性好,很容易构建多官能团的分子.因此,研究MBH酯的反应比MBH反应更有应用价值,受到有机化学工作者的广泛关注.目前,MBH酯作为底物的烯丙基烷基化反应已有不少报道,所使用的催化剂主要有有机小分子催化剂和金属与膦配体的催化体系.常用的小分子催化剂包括金鸡纳碱氢化奎尼定1,4-(2,3-二氮杂萘)二醚[(DHQD)2PHAL][1~3]、金鸡纳碱衍生物[4,5]、氢化奎宁蒽醌-1,4-二甲氨基)二醚[(DHQD)2AQN][6~10]、氢化奎宁-2,5-二苯基-4,6-嘧啶二甲醚[(DHQD)2PYR][11]、β-6′-羟基异辛可宁(β-ICD)[12]、奎尼丁[13]、奎尼丁衍生物[14]和有机膦小分子催化剂[15~19]. 常见的金属与膦配体催化体系有金属铱、钯与单膦配体[20~26]和金属钯与双膦配体[27~29].
本文首次研究了过渡过金属铱与亚磷酰胺和膦配体催化的MBH乙酸酯与3-苯基吲哚酮类化合物的烯丙基取代反应,以良好至优秀收率获得了一类具有新结构的3,3-二取代的吲哚酮类化合物.
1 实验部分
1.1 试剂与仪器
靛红、丙烯酸乙酯、乙酸酐、乙酸、无水硫酸钠、无水碳酸钠、1,4-二氮杂二环[2.2.2]辛烷(DABCO)、三乙胺、苯甲醛、苄溴、苯基溴化镁、苯甲醛类化合物、1,8-二氮杂二环十一碳-7-烯(DBU)、不同取代的靛红类化合物和金属试剂等,均为阿达玛斯试剂有限公司或其它市售分析纯试剂.
1,4-二氧六环(Dioxane)和乙二醇二甲醚(DME)经钠回流干燥除水;二氯甲烷经CaH2回流、蒸馏后脱气使用;四氢呋喃(THF)和甲苯均经金属钠回流,以二苯甲酮作指示剂,蒸馏后脱气使用.
Bruker AVANCE III型核磁共振波谱仪和APEX 47e ESI FT-ICR质谱仪,德国布鲁克公司;1200型高效液相色谱(HPLC)分析仪,美国安捷伦科技有限公司.
1.2 实验过程
1.2.1 MBH乙酸酯的合成 MBH乙酸酯(2a~2i)参考文献[27]方法合成.
1.2.2 3-苯基吲哚酮类化合物的合成 吲哚酮类化合物(1a~1f)参考文献[30]方法合成并纯化.
1.2.3 吲哚酮类化合物与MBH乙酸酯的烯丙基烷基化反应 反应过程如Scheme 1所示.以化合物3a的催化合成为例:将烘干后的20 mL反应管和橡胶塞带进手套箱,分别称取5.04 mg金属铱络合物[Ir(COD)Cl]2(摩尔分数5%)和5.40 mg配体L6(摩尔分数10%),再加入1 mL乙腈溶剂,用橡胶塞密封好反应管,从手套箱中取出反应管,固定在磁力搅拌器上,室温下搅拌1 h.

Scheme 1 Synthetic routes of compounds 3a—3r
称取44.8 mg吲哚酮类化合物1a(0.15 mmol)、44.7 mg MBH乙酸酯2a(0.18 mmol)和146.6 mg Cs2CO3(0.45 mmol)加入至聚乙烯离心管中,与上述反应管一起放入手套箱中,然后将离心管中的底物和碱加入到反应管中,再加入4 mL乙腈,密封好反应管,从手套箱中取出,放到-30℃低温反应器中反应.以薄层层析法检测反应进程,反应完全后取出反应管,减压浓缩除去溶剂.再经柱层析纯化[展开剂为石油醚-乙酸乙酯(体积比20∶1)]得到无色油状产物.化合物3b~3r也按此方法合成.目标化合物3a~3r的理化性质结果及核磁共振波谱数据分别如表1和表2所示,核磁共振波谱(图S1~图S54)见本文支持信息.

Table 2 1H NMR and13C NMR data of compounds 3a—3r

Continued
2 结果与讨论
2.1 金属催化剂的筛选
不对称烯丙基烷基化反应的常用金属为Pd和Ir等过渡金属,与手性配体一同使用进行催化[20~29].首先,选用DBU为碱,DME为溶剂,联萘类型的亚磷酰胺为配体[31]对金属源进行筛选,结果如表3所示.可见,不同的金属对反应影响很大,其区域选择性有很大差别.从几种不同的钯源对反应的催化效果可知,大部分钯源(表3中Entries 1~6)催化生成的主要产物为此类底物烯丙基化反应常见的反应产物4a,其产率最高达50%(表3中Entry 6),说明金属钯与亚磷酰胺配体催化体系对该反应有一定的催化效果,生成的主要产物为MBH乙酸酯烯丙基被取代的产物4a.通过对产物4a进行HPLC分析,发现其没有对映选择性,但反应有一定的非对映选择性.当将金属钯换成铱时,反应位点发生改变,生成的主要产物变为3a,这样的区域选择尚未见报道,且3a产率明显提高,达到88%(表3中Entry 7);反应时间也显著缩短,原料只需要20 h就几乎转化完全.对产物进行柱层析纯化,然后经HPLC分析,发现产物3a也无对映选择性.
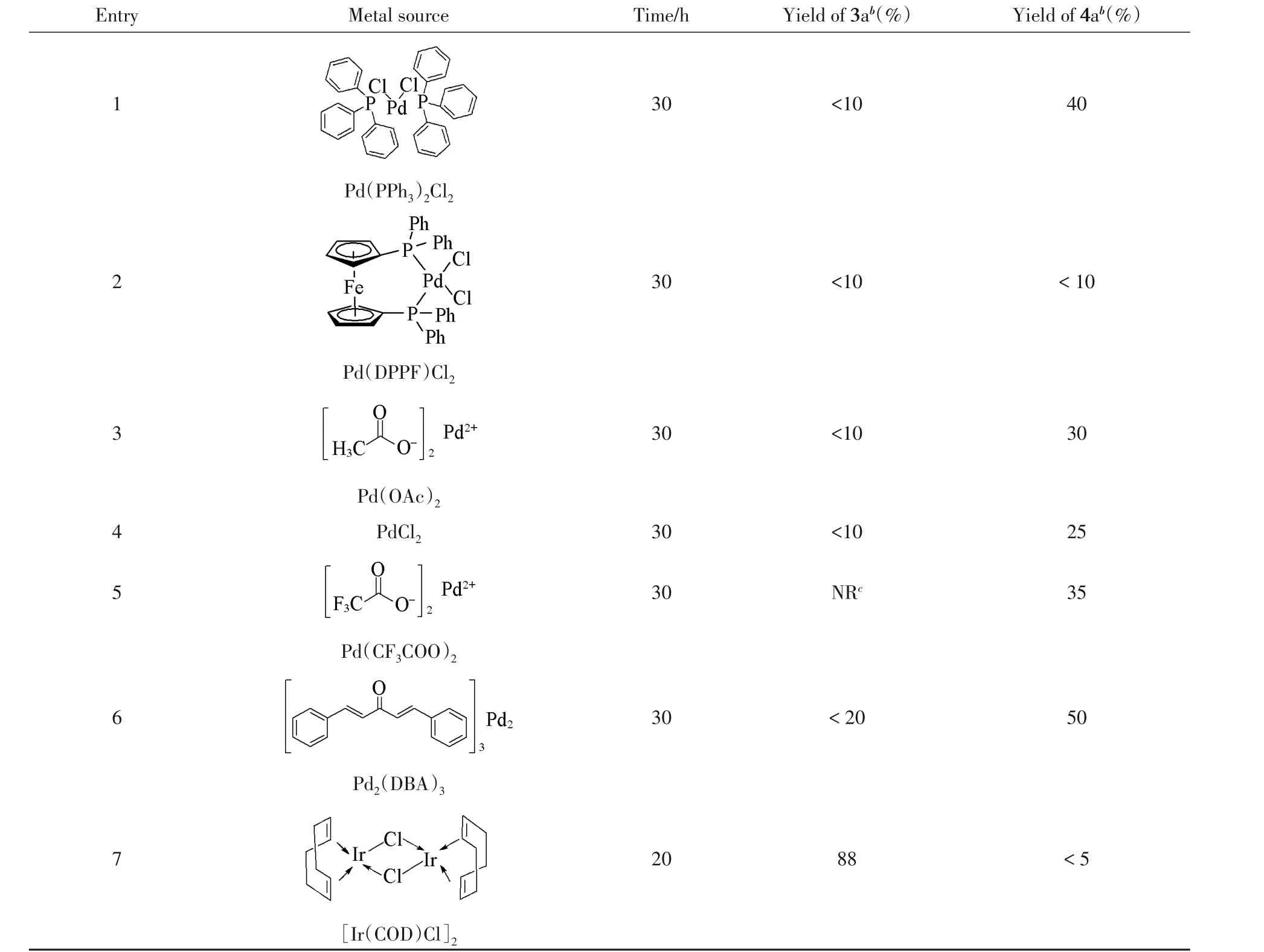
a..Reaction conditions:1a(0.15 mmol,100%),2a(0.18 mmol,molar fraction 120%),DME(4 mL),metal source(molar fraction 5%),ligand(molar fraction 10%),DBU(molar fraction 300%),-30℃;b.isolated yield;c.NR:no reaction.
2.2 反应条件的优化
利用铱与亚磷酰胺配体催化此反应,考察溶剂对反应的影响,结果如表4所示.可见,催化反应的溶剂效应明显,当用二氯甲烷(DCM)和二氯乙烷(DCE)作为溶剂时,产率仅有30%(表4中Entry 1)和40%(表4中Entry 3);在极性溶剂中此反应体系的催化效果较好,可以显著提高反应的收率;乙腈为溶剂时产率最优,达到90%(表4中Entry 5),但经HPLC分析发现产物无对映选择性.
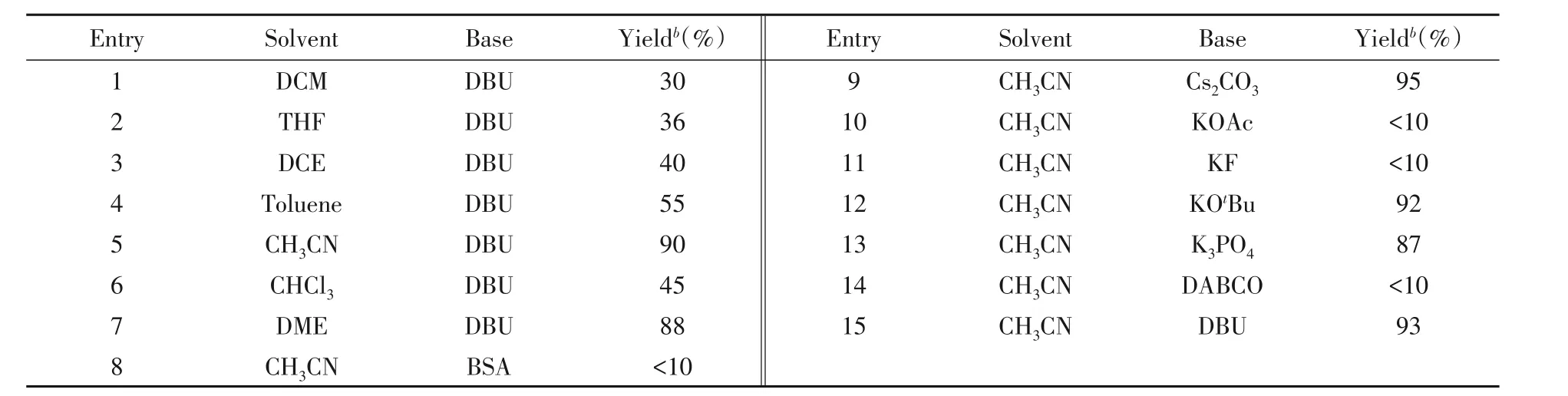
a..Reaction conditions:1a(0.15 mmol,molar fraction 100%),2a(0.18 mmol,molarfraction 120%),solven(t4 mL),[Ir(COD)Cl](2molar fraction 5%),ligand(molar fraction 10%),base(molar fraction 300%),−30℃,20 h;b.isolated yield.
由表4可知,碱对反应的产率影响很大.以N,O-双三甲硅基乙酰胺(BSA),KOAc,KF和DABCO作为碱时,反应产率很低,几乎不反应(表4中Entries 8,10,11和14);Cs2CO3作为碱时反应产率最高(表4中Entry 9),并且反应时间相对较短;KOtBu,K3PO4和DBU作为碱时,产率最低也达到87%(表4中Entries 12,13和15).表明碱的强弱对底物的亲核性有很大影响,中等强度的无机碱或有机碱更适合此反应.HPLC分析发现,产物仍无对映选择性.可能是新的催化体系在催化过程中没有手性控制.通常,在不对称合成中配体的结构对产物的对映选择性影响很大,对配体结构进行小的修饰,结果可能不同.因此,继续对不同结构的亚磷酰胺配体及其它类型的配体进行筛选,以获得理想的催化效果.
2.3 配体的筛选
不同配体的筛选结果如表5所示.为确保底物转化完全,将反应时间延长至25 h.结果表明,轴手性联苯类型的亚磷酰胺配体[32,33](L1~L4,L7,L9~L11和L13)和联萘类型的亚磷酰胺配体(L5,L6,L8和L12)都有很好的催化活性,产率最低可达90%(表5中Entries 1~13);其中,使用手性丁二醇(L1,L2,L7,L9~L11和L13)和戊二醇(L3)桥连联苯类型的亚磷酰胺配体时,产率最低也达92%(表5中Entries 1~3,7,9~11和13);当配体氨基部位有较大位阻时(L8),催化效果稍有下降,产率为90%(表5中Entry 8);以R-(+)-1,1′-联萘-2,2′-二苯膦(L14)作为配体时,产率为97%(表5中Entry 14);使用3,3-二甲基取代的联萘亚磷酰胺配体(L12)时,产率为95%(表5中Entry 12);使用氨基部位为2个甲基的联萘配体(L6)时,产率最高达98%(表5中Entry 6);使用N-P三齿配体(L16)时,产率为91%(表5中Entry 16);使用其它的含磷配体(L15和L17)时,产率均为93%(表5中Entries 15和17).对不同配体催化合成的产物进行HPLC分析,仍未发现产物具有对映选择性.
综上所述,反应的最优条件为以[Ir(COD)Cl]2(5%)和L6(10%)作为催化剂,Cs2CO3(300%)为碱,CH3CN(4 mL)为溶剂,于−30℃反应25 h.
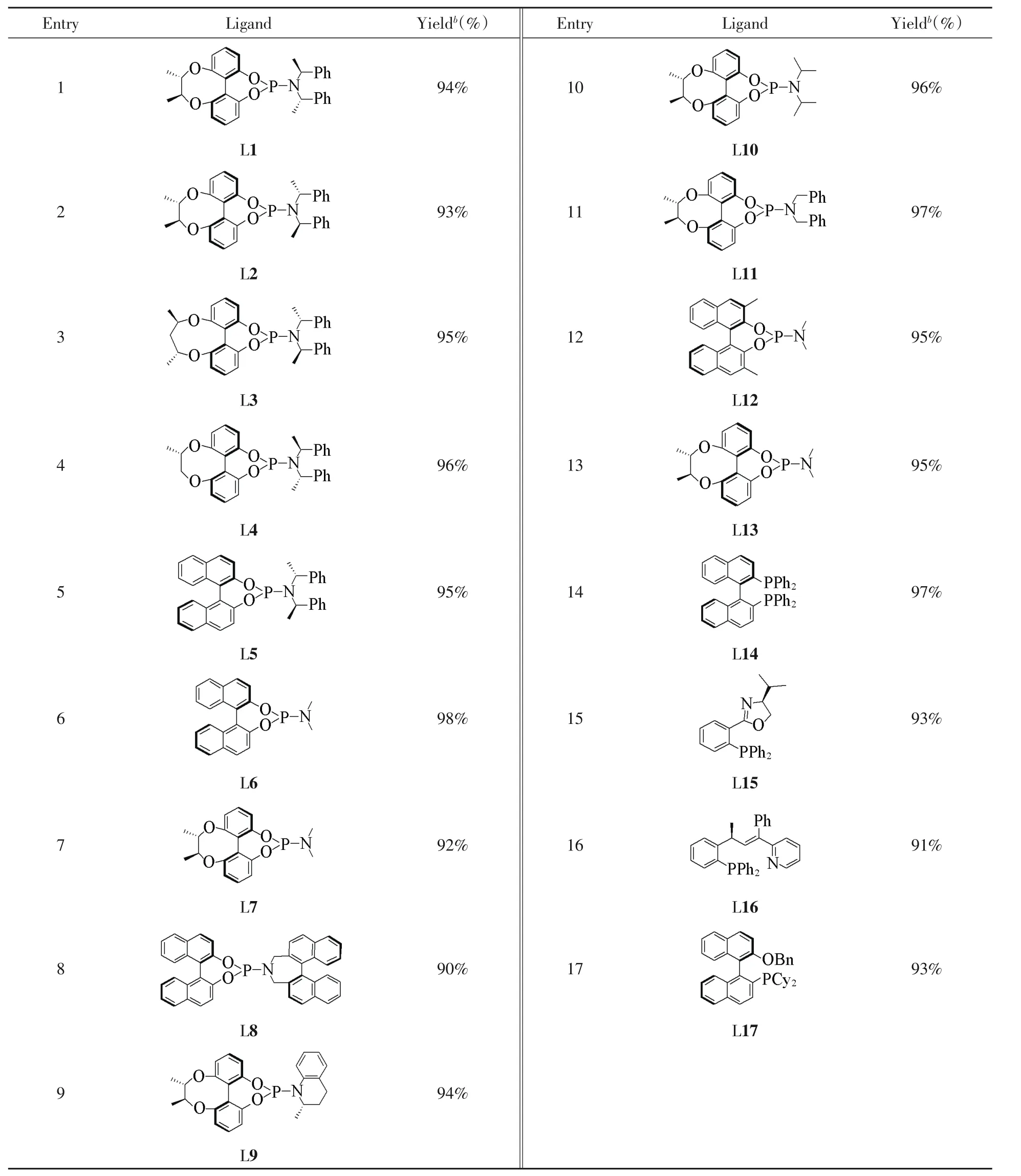
Table 5 Influence of ligand on the yield of the reactiona
2.4 底物的拓展
在上述最优反应条件下,对底物的范围进行了拓展,结果如表1所示.当3-苯基吲哚酮的C5位为给电子基(甲基和甲氧基,3o,3p)时,产率比C5位具有吸电子基(氟和氯,3m,3n)时稍高;当C6位和C7位为氟原子取代时(3q,3r),产率有所下降,特别是C7位为氟原子时产率显著下降为84%.表明当吲哚酮苯环上具有吸电子基,尤其是C6和C7位为吸电子基时,会导致产率下降.当3-苯基吲哚酮的C3位苯环上取代基为较强给电子基,如4-甲氧基和2,5-二甲氧基时,产率较高(3k,3l).当MBH乙酸酯苯环的C2位为硝基时,产率为93%(3b),表明C2位为吸电子基时较不利于反应的进行.当MBH乙酸酯苯环的C3位为给电子基甲基时,产率为96%(3i);而C3位为吸电子基团,如氟、氯和溴时,产率最高只达94%(3d~3f),可知C3位具有吸电子基团时不利于反应的进行,导致产率下降.当MBH乙酸酯苯环的C4位为三氟甲基和氰基取代时,产率分别为92%和93%(3g,3h);而当苯环的C3和C4位为氯原子时,产率为91%(3c);当芳杂环为噻吩时,产率为94%(3j),表明无论是3-芳基吲哚酮或是MBH乙酸酯,苯环上带有吸电子基团时比带有给电子基团时产率都稍有降低,但总体影响不大,产率最低亦可达84%(3r).
3 结 论
研究了一种新的催化方法,通过过渡金属铱和亚磷酰胺配体催化Morita-Baylis-Hillman(MBH)乙酸酯与3-苯基吲哚酮类化合物进行烯丙基取代反应,高效合成了一类以往烯丙基取代反应中未见报道的具有新结构的3,3-二取代吲哚酮类化合物.铱催化的区域选择明显不同于钯催化的反应.反应的底物普适性较好,在优化的催化反应条件下,具有不同类型取代基的底物反应的产率最低为84%,最高可达98%.底物取代基的电性对反应产率影响不大,一些其它类型的双膦和单膦配体对催化反应也有较好的效果.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20200519.