酿酒酵母NST1基因敲除菌的构建及其对抗氧化的影响
2020-10-15王梦飞焦宏亮王金鹏
朱 红,王梦飞,焦宏亮,王金鹏,周 华,蔡 恒
(南京工业大学 生物与制药工程学院,江苏 南京 211800)
自由基的生成在老龄化相关的疾病中起着核心作用[1-3]。自由基作为生物体的基本保护机制,能够参与各种代谢信号途径的调节,传递生命活动所需的能量,也可作为吞噬细胞消灭细菌或病原体的主要介质。此外,自由基还影响基因的转录和表达,参与重要生命活动的调节[4],但自由基尤其是活性氧(ROS)的过量生成则会造成机体内氧化/还原的失衡,促进氧化应激(oxidative stress)的发生,致使细胞结构、脂类、蛋白质和遗传物质等遭到破坏,进而引起细胞凋亡,诱发疾病。
ROS主要有2种来源:一种是内源性来源,如线粒体、NADPH氧化酶和过氧化酶体等,其中有超过80%活性氧是由于线粒体呼吸链氧化磷酸化过程中电子泄漏引起的[5-6];另一种外源性来源,如外源辐射、病原体或重金属的刺激等[7]。由ROS引起的冠心病、糖尿病和肿瘤等疾病的病理机制研究已取得较大进展[8-13]。大脑对ROS尤为敏感[14],过多的ROS刺激更易诱发帕金森病和阿尔茨海默病等神经系统退行性病变发生[15-17]。因此,针对ROS生成或ROS损伤预防的研究具有重要意义。酵母体内衰老代谢机制与人体细胞衰老机制相似,并且酵母基因组与人类基因组高度同源,其生长代谢规律简单独特,单倍体状态的可操作性更有利于进行多操作实验,因而成为细胞抗衰老和疾病研究的最优模型[18]。
酵母作为一种需氧生物,常常受到高水平活性氧的毒害,已进化出一套较为完整的抗氧防御系统,主要包括非酶系防御系统和酶系防御系统两大类。酶系防御系统主要由过氧化氢酶、超氧化物歧化酶等组成,非酶系防御系统则主要由谷胱甘肽、硫氧还蛋白等组成,其中谷胱甘肽最为重要。真核生物细胞壁完整性信号通路(CWI)能检测真菌细胞在受到高渗、热刺激、抗真菌药物等环境刺激时产生的胞壁胁迫信号并及时作出应答。Goossens等[19]研究发现NST1基因的敲除增强了酵母的耐盐能力,但其具体机制未知。Leng等[20]研究发现NST1基因编码的蛋白作为一种高渗透压甘油响应途径(HOG)的“补充途径”加强了CWI途径,从而提高酵母细胞的耐热能力。这对研究CWI途径在抗氧化影响方面提供了思路。
因此,本研究中,笔者采用Cre-LoxP系统、Kanr抗性标记和同源重组技术敲除NST1基因,构建酿酒酵母NST1基因缺失菌株,以分析NST1基因对酵母抗氧化能力的影响。
1 材料与方法
1.1 材料
1.1.1 菌株、试剂及引物
酿酒酵母BY4741(S.cerevisiaeBY4741,MATahis3Δ1leu2Δ0met15Δ0ura3Δ0)、质粒pUC18保藏于笔者所在实验室;质粒pUG6在大肠杆菌中具有氨苄青霉素抗性,质粒pUG6带有Kanr抗性基因,且两端带有loxp位点,广西大学杜丽琴教授惠赠;质粒pYES2由天津科技大学惠赠,在大肠杆菌中具有氨苄青霉素抗性,能利用半乳糖诱导蛋白在酵母中表达。
rTaq酶、HindⅢ、SacⅠ、T4连接酶、PrimeSTAR@Max DNA Polymerase、DNA Marker DL2000、dNTP Mixture、Agarose Gel DNA Purification、5×PrimeSTAR Buffer(Mg2+plus)、质粒小量提取试剂盒、小量琼脂糖凝胶DNA回收试剂盒及相关限制酶,TaKaRa 公司;基因提取试剂盒、G418,生工生物工程(上海)股份有限公司;PCR 仪,Biometra公司;电转仪、凝胶成像系统,Bio-Rad 公司;高速冷冻离心机,Eppendorf公司;荧光显微镜,德国莱卡公司;超低温冰箱,Sanyo公司;培养箱,上海志成生物公司;电泳仪,北京六一生物科技有限公司;其余试剂均为进口或国产分析纯。
根据酶母基因组数据库(SGD)数据库的NST1基因(S000005035)和 GenBank 的质粒序列设计引物,敲除引物,命名为QC-F、QC-R,基因NST1验证引物为 A、Yz-a、Yz-b和B。根据HindⅢ、SacⅠ酶切位点设计NST1回补引物HB-F、HB-R。引物均由南京金斯瑞有限公司合成,所涉及的引物及其序列见表1。

表1 构建菌株引物
1.1.2 培养基
YPD培养基:酵母膏5 g/L、蛋白胨10 g/L、葡萄糖20 g/L;自然pH,去离子水定容至1 000 mL,灭菌待用,微量氨苄青霉素(Amp)。
YPD+G418培养基:每50 mL培养基加入500 μL质量浓度为10 mg/mL的G418母液,终质量浓度为100 μg/mL,用于酵母转化子的筛选。
半乳糖诱导表达培养基(g/L):酵母膏5、蛋白胨10、半乳糖20。
SC-Ura培养基:每100 mL培养基中含20 g/L葡萄糖,6.7 g/L Yeast Nitrogen Base(YNB),L-Leu、L-His和L-Met(质量浓度为10 mg/mL)各1 mL,固体培养基需加入15~20 g/L琼脂。
LB培养基(g/L):酵母提取物5、胰蛋白胨 10、NaCl 10、琼脂 20(配制固体培养基)。用于大肠杆菌的培养和保藏。
LB+Amp培养基:每100 mL培养基加入100 μL的50 mg/mL的Amp母液,终质量浓度为50 μg/mL,用于含质粒大肠杆菌的培养与提取。
LB蓝白筛培养基:每100 mL培养基加入质量浓度为 24 mg/mL IPTG、100 mg/mL Amp母液各100 μL,20 mg/mL X-gal 母液200 μL。
磷酸缓冲液(PBS)(g/L):KCl 0.2、KH2PO40.24、NaCl 8.0、Na2HPO41.44;调节pH至7.4。
以上培养基均需在高压蒸汽灭菌锅中121 ℃灭菌20 min,葡萄糖单独灭菌;G418与Amp须在培养基降至合适温度后加入。
1.2 方法
1.2.1 含G418抗性基因(kanr)及NST1同源臂PCR扩增
所用的大肠感受态细胞及质粒转化参照分子克隆中电击转化法操作。利用碱裂解法从大肠杆菌中提取质粒pUG6,以其为模板,以 QC-F、QC-R 为引物进行 PCR扩增,获得同源臂NST1基因敲除组件。扩增条件如下:95 ℃ 5 min;95 ℃ 30 s;72 ℃ 30 s;72 ℃ 2 min;72 ℃ 10 min。取 2 μL PCR产物进行0.8%琼脂糖凝胶电泳检测。该 PCR产物即为含G418抗性基因(kanr)及NST1同源臂目的片段,片段大小在1 800 bp左右。
1.2.2 酿酒酵母电转感受态细胞制备
将S.cerevisiae单菌落接种到50 mL YPD液体培养基中,30 ℃、200 r/min振荡培养过夜。按体积分数2%接种量将种子液接种至100 mL YPD培养基中,接种2瓶。再于30 ℃、250 r/min条件下振荡培养3~5 h。将培养的菌液转移到冰预冷的离心管上,放置冰上30 min,并将离心机预冷至4 ℃,3 000 r/min离心15 min,去上清;用200 mL冰预冷的超纯水洗涤重悬菌体,4 ℃、3 000 r/min离心15 min,洗涤2次;用100 mL冰预冷的1 mol/L山梨醇洗涤重悬菌体,3 000 r/min离心15 min,去上清,再用100 mL冰预冷的山梨醇洗涤重悬菌体,4 ℃、3 000 r/min离心15 min,倒去上清,用残留的山梨醇将菌体溶解,不需加入新的山梨醇溶解。溶解后的菌体,每管50 μL分装至冰预冷的1.5 mL EP离心管中,置于-80 ℃保存。
1.2.3NST1基因敲除菌构建
将含G418抗性(Kanr)及NST1同源臂 PCR产物(约 400 ng)加入新制备的100 μL感受态细胞中,混匀,转移至2 mm电转杯预冷5 min,并使用Eppendorf电转仪进行电转化(电击参数:电压1.5 kV,电容25 μF,电阻400 Ω,电击间距2 mm)。电转后立即加入1 mL预冷山梨醇复苏,混匀,8 000 r/min离心1 min,倒去部分上清,留下100 μL浓缩涂布于含200 μg/mL G418 的 YPD筛选平板,30 ℃培养2~3 d观察是否有转化子。
1.2.4 酿酒酵母转化子的筛选与鉴定
将平板上出现的单菌落挑选至含200 μg/mL G418 的YPD 液体培养液中,30 ℃培养过夜后,8 000 r/min离心2 min收集菌体,使用上海生工生物工程有限公司提供的试剂盒提取重组菌DNA,并以获得的基因组为模板,用 Taq DNA 聚合酶,引物对A/Yz-a、B/Yz-b 进行PCR验证NST1基因敲除菌,验证成功敲除菌命名为nst1Δ。酵母基因提取参照试剂盒说明书进行操作。
1.2.5 回补菌株nst1Δ-NST1的构建
以BY4741 基因组为模板,HB-F、HB-R为引物PCR扩增出带酶切位点NST1片段。提取质粒 pUC18和pYES2,核酸内切酶HindⅢ和SacⅠ分别处理质粒和PCR扩增的NST1基因,T4 DNA连接酶连接pUC18载体和基因片段,构建载体 pUC18-nst1,电转转入大肠感受态细胞。送检测序后,HindⅢ、SacⅠ双酶切,切胶回收。T4 DNA连接酶连接回收片段与pYES2载体,电转导入敲除菌nst1Δ酵母感受态细胞中,涂布于尿嘧啶营养缺陷平板,3 d后挑取转化子,利用pYES2通用引物验证。
1.2.6 不同H2O2浓度的稀释点滴实验
将验证成功的重组菌接种至含100 μg/mL G418的10 mL YPD中,180 r/min、30 ℃摇床培养过夜制备种子液,离心后将细胞重悬至相似的OD600,然后分别稀释至10-1、10-2、10-3、10-4和10-5,取3 μL分别点滴在普通YPD及含有4 mmol/L H2O2浓度平板上(半乳糖诱导),倒扣培养3 d,观察生长情况。
1.2.7NST1基因敲除菌株生长速率比值分析
1.2.8 ROS测定
活性氧检测试剂盒(南京建成公司)测定酵母胞内ROS水平。将BY4741 和nst1Δ单菌落分别接种于普通YPD培养基中,180 r/min、30 ℃摇床过夜培养,隔夜培养物稀释于新鲜YPD中,调整OD600为0.1,再180 r/min、30 ℃摇床培养4 h后,分别加入4 mmol/L H2O2进行氧化处理2 h,离心后将PBS洗涤2次,PBS重悬,加入适量2′,7′-二氯荧光黄双乙酸盐(DCFHDA)染剂,30 ℃孵育40 min,PBS洗涤重悬,取200 μL于荧光显微镜中观察。
1.2.9 荧光定量PCR测定
酵母细胞总RNA的提取按照TaKaRa试剂盒说明在4 ℃条件下提取。Q-PCR反应液的配制(总体积20 μL):5×PrimeScript RT Master Mix,4 μL;模板RNA,1 μL;去DEPC水,15 μL。反应液配制均在冰上进行。轻轻混匀后,在PCR仪上按下列条件进行反转录反应:37 ℃ 15 min;85 ℃ 5 s;4 ℃10 min。
荧光定量PCR反应体系。2×qPCR Master Mix,10 μL;正向引物,0.4 μL;反向引物,0.4 μL;RT反应液(cDNA),2 μL;dd H2O,7.2 μL。
荧光定量PCR扩增条件。扩增曲线:95 ℃ 5 min 1 循环;95 ℃ 5 s,60 ℃ 31 s 40循环。熔解曲线:95 ℃ 15 s,60 ℃ 30 s,95 ℃ 15 s。
所需荧光定量PCR引物为RLM1-F:GCAAACTGGTGCTCAAGG;RLM1-R:GGTCCCGAAGTAGGAAAGG。
2 结果与讨论
2.1 NST1基因敲除序列制备
PCR扩增G418抗性基因的同时引入NST1基因同源臂,用于后续基因同源重组。以 QC-F、QC-R 为引物,利用 PCR 获得带有 2 个NST1同源臂和kanr筛选标记基因片段,分析结果见图1。由图1可以发现,实验得到片段大小与理论预计(1 800 bp)一致,该PCR 产物即为NST1基因敲除序列组件。
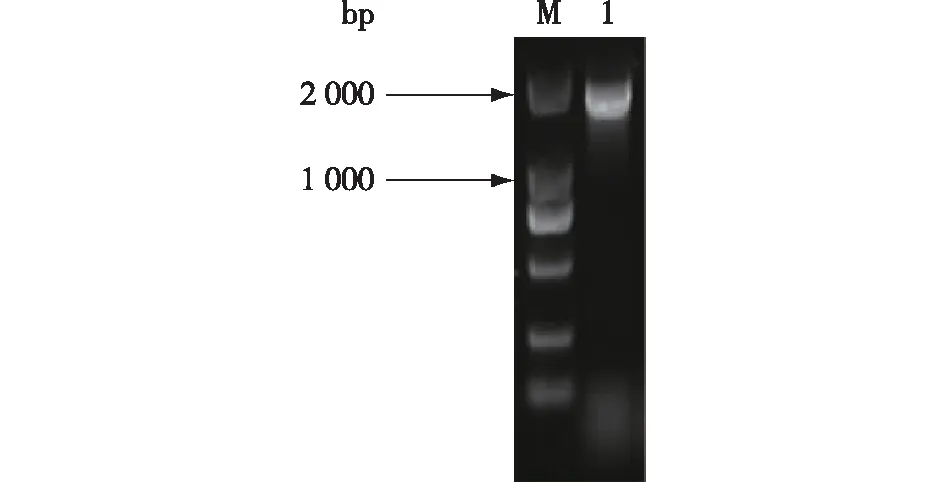
M—DL2000; 1—nst1Δ PCR产物
2.2 重组敲除菌株nst1Δ构建与验证
采用电击转化法将NST1基因敲除序列组件转化至酿酒酵母中。由于NST1基因敲除序列组件中包含异源显性的kanr标记,将构建好的序列组件转化至酵母细胞后其两端与酵母基因组同源的序列将引发同源重组替换,最终以loxp-Kanr-loxp取代基因组中的NST1从而赋予转化子G418(遗传霉素)抗性。2~3 d后可在含G418的平板上获得转化子(图2)。

图2 NST1基因敲除序列组件转化
提取转化子基因组,以A/Yz-a、B/Yz-b为引物,进行PCR扩增。正确整合了敲除组件的细胞使用引物A与YZ-a配对可得到200 bp左右产物,使用引物B与YZ-b可得到800 bp左右产物,如图3所示。由图3可知,转化子验证结果与理论值一致,并且测序结果亦显示NST1基因敲除成功。
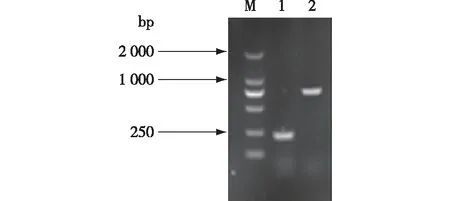
M—DL2000; 1—A-YZ-a PCR产物; 2—YZ-b-B PCR产物
2.3 回补菌株nst1Δ-NST1的构建与验证
将连接载体pUC18-nst1 电转转入大肠感受态细胞中,涂布于LB蓝白筛培养基,过夜挑取转化子,提取质粒,核酸内切酶双酶切所提取质粒,电泳结果如图4(a)所示。将阳性克隆的片段送检测序,验证成功后,酶切胶回收构建pYES2-nst1载体,并双酶切后电泳分析,结果如图4(b)所示。将正确构建的pYES2-nst1载体电转导入nst1Δ敲除菌感受态,提取转化子基因组,pYES2通用引物PCR扩增,进行电泳分析,结果如图4(c)所示。由图4可知,回补菌株nst1Δ-NST1构建成功。

M—DL5 000; 1—pUC18-NST重组载体酶切验证;M′—DL10 000;1′—pYES2-NST1重组载体酶切验证; M″—DL2 000;1″—pYES2通用引物验证
2.4 重组菌株不同H2O2浓度的稀释点滴实验
将BY4741、nst1Δ敲除菌及nst1Δ-NST1回补菌接种于10 mL半乳糖诱导培养基中,30 ℃过夜培养,离心收集菌体,加入PBS液使菌悬液OD600为1.0,进行稀释梯度点滴实验,依次吸取3 μL点滴于测试平板上,30 ℃倒扣培养3 d,观察菌落生长情况,结果如图5所示。由图5可知,与对照菌BY4741相比,敲除菌对H2O2浓度更为敏感,在含4 mmol/L H2O2平板上,生长抑制明显,回补之后生长有所恢复,说明NST1基因的敲除降低了酵母的抗氧化能力。

图5 BY4741/nst1Δ/nst1Δ-NST1在不同H2O2浓度平板上的表型实验(n=3,3 d)
2.5 氧化应激下重组菌株生长速率比值的分析
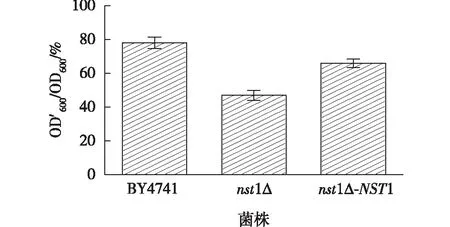
图6 BY4741、nst1Δ与nst1Δ-NST1在4 mmol/L H2O2下生长速率比
从图6可知,在4 mmol/L H2O2YPD液体中,对照菌的生长速率比在78%左右,而敲除菌则下降至47%,与对照菌相比,敲除菌的生长明显受到抑制,这结果表明NST1基因敲除确实降低了酵母抗氧化性能。Goossens等[19]研究发现,NST1基因的敲除能够增强酵母耐盐能力。这可能是由于在高渗透压胁迫下,酵母需要更多甘油作为一种可溶性有机溶剂,以维持胞内外渗透压平衡[21],所以,NST1基因的敲除反而相对增强HOG途径,表现出更强的耐盐性。在氧化应激下,细胞激活CWI途径用以维持细胞壁完整性,NST1的缺失导致CWI补充途径受到阻碍,增强了细胞的氧敏感性。
2.6 荧光显微镜检测胞内ROS
利用活性氧检测试剂盒来测定对照菌和重组菌胞内ROS水平。试剂盒采用荧光探针2′,7′-二氯荧光黄双乙酸盐(DCFH-DA)进行活性氧检测,DCFH-DA本身没有荧光,可以自由穿过细胞膜,进入细胞内后,可以被细胞内的酯酶水解生成DCFH。而DCFH不能通透细胞膜,从而使探针很容易被装载到细胞内。细胞内的活性氧可以氧化无荧光的DCFH,生成有荧光的DCF。对实验组和对照组进行处理后,荧光显微镜下观察两种菌在不同培养条件下的ROS水平,结果如图7所示。
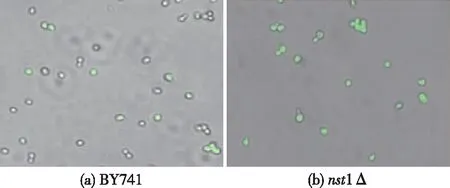
图7 在4 mmol/L H2O2条件下胞内ROS水平
由图7可知,经H2O2处理后,对照菌只有少许细胞胞内含有ROS,而敲除菌几乎是所有的细胞胞内存在ROS。由此可见,NST1基因的敲除增加了酵母胞内ROS水平。
2.7 荧光定量PCR测定CWI途径下游基因转录水平
NST1基因编码的蛋白作为一种HOG途径的“补充途径”加强了CWI途径,当细胞受到氧化应激时,NST1可加强CWI途径下游基因Slt2磷酸化状态,促进下游因子RLM1的表达,维持细胞壁的完整性。为此,采用Q-PCR技术对CWI途径中RLM1进行了基因转录水平测定,结果如图8所示。

图8 基因RLM1转录水平
由图8可看出,在正常培养条件下,对照菌RLM1的表达水平上升到了423.3%,而敲除菌上升幅度相对变小,只有215.6%。由此,可以推测NST1的缺失影响了酵母CWI途径下游RLM1基因的转录表达,其控制的大量涉及细胞壁合成的基因。细胞壁完整性得不到维持可能是敲除菌抗氧能力降低的主要因素。
3 结论
以酿酒酵母模式菌株BY4741为出发菌株,通过同源重组技术对酵母NST1基因敲除,命名为nst1Δ,并考察其抗氧化性能。与对照菌BY4741相比,敲除菌对H2O2浓度更为敏感,在4 mmol/L H2O2YPD液体培养中,敲除菌生长速率受到明显抑制。胞内ROS检测结果亦说明NST1基因敲除增加了胞内ROS水平。NST1作为“补充途径”从HOG途径方面加强CWI途径以维持细胞壁完整性,实验结果表明NST1基因的缺失降低了RLM1基因的表达水平。
